Elektrochemische Doppelschicht
Elektrochemische Doppelschicht, elektrolytische Doppelschicht oder kurz Doppelschicht sind gebräuchliche Namen für Grenzschichten, an denen sich elektrisch getrennte geladene Schichten gegenüberstehen.
Aufbau
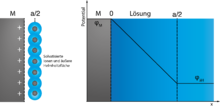
In der Regel versteht man unter der elektrochemischen Doppelschicht die Phasengrenze zwischen einem Elektronenleiter (der Elektrode) und einem Ionenleiter (dem Elektrolyten). Auch an der flüssig-flüssig-Phasengrenze nicht mischbarer Elektrolyten tritt eine „Doppelschicht“ auf. Typischerweise stehen sich an der Phasengrenze im geladenen Zustand zwei Ladungsschichten gegenüber, die – wie in jedem Kondensator – entgegengesetzte Vorzeichen tragen. Die entladene Doppelschicht trägt auf den Elektroden das so genannte Nullladungspotential, bei dem die Metallseite ungeladen ist und auch die Lösungsseite keine Nettoladung trägt. Die „Dicke“ der geladenen Schichten, d.h. die mittlere Ausdehnung senkrecht zur Oberfläche, beträgt in Metallen etwa 0,1 nm, in der Lösung 0,1 bis 10 nm; sie wird durch die Debye-Länge beschrieben. In der Lösung ist sie von der Beweglichkeit der Ionen und von der Konzentration der Lösung abhängig, im Metall vor allem von der Elektronendichte, da die Atomrümpfe in festen Elektroden nicht beweglich sind.
Historische Entwicklung der Vorstellungen zur Doppelschicht
Helmholtz-Modell
Hermann von Helmholtz stellte die ersten Überlegungen und Untersuchungen zu Doppelschichten an, siehe dazu den Artikel Helmholtzschicht.
Gouy-Chapman-Modell
Das frühe Helmholtz-Modell beschrieb lediglich eine konstante Differenzialkapazität unabhängig von der Ladungsdichte und nur abhängig von der Dielektrizitätskonstante und der Dicke der Doppelschicht. Aber dieses Modell ist nur eine gute Grundlage für die Beschreibung der Ladungstrennung. Sie berücksichtigt nicht wichtige Faktoren wie Diffusion bzw. Vermischung von Ionen im Lösungsmittel, die Möglichkeit der Adsorption von Ionen an der Oberfläche der Elektrode und die Wechselwirkung zwischen Dipolmomenten im Lösungsmittel und in der Elektrode.
Deshalb wurde 1910 von Louis Georges Gouy und 1913 von David Leonard Chapman die Theorie von Helmholtz weiterentwickelt. Sie gingen von einer thermischen Bewegung der Gegenionen im Elektrolyten aus, die zur Bildung einer über mehrere Moleküllagen ausgedehnten diffusen Schicht führte, der sogenannten Gouy-Chapman-Doppelschicht, die spannungsabhängig ist und auch noch von der Konzentration der Ionen abhängt. In diesem Modell wird die Ladungsverteilung von Ionen im Elektrolyten als eine Funktion der Entfernung von der Metalloberfläche verstanden und kann mit der Boltzmann-Statistik beschrieben werden. Das bedeutet, dass das elektrische Potential exponentiell von der Oberfläche der Flüssigkeit abnimmt.
Stern-Modell
Das Gouy-Chapman-Modell versagt jedoch bei stark geladener Doppelschicht. 1924 vereinigte Otto Stern die Vorstellungen von Helmholtz mit der von Gouy und Chapman. In seinem Modell der Doppelschicht setzt sich die Schicht im Elektrolyten aus einer starren und einer daran anschließenden diffusen Schicht zusammen, so dass sich das Modell der Stern-Doppelschicht ergibt. Dieses Modell berücksichtigt die Tatsache, dass Ionen eine endliche Größe haben. Folglich ist die größtmögliche Annäherung der Ionen an die Elektrode in der Größenordnung des Ionenradius.
Grahame
Aber das Modell von Stern hat noch einige Einschränkungen, beispielsweise sind die Ionen nur als Punktladung modelliert, wobei die einzige signifikante Wechselwirkung in der diffusen Schicht die einer elektrischen Ladung ist, außerdem wird die Permittivität über die Doppelschicht als konstant vorausgesetzt ebenso wie die Viskosität des Elektrolyten.
_NT-int.svg.png)
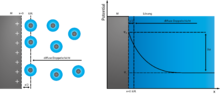
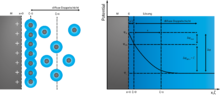
Deshalb modifizierte David C. Grahame 1947 das Stern-Modell unter Berücksichtigung des Lösemittels und der Solvatation sowie der spezifischen Adsorption. Im Grahame-Modell wird die Helmholtz-Schicht zur festen Phase hin durch die innere Helmholtz-Fläche (englisch inner Helmholtz plane, IHP) begrenzt. Sie verläuft durch die Mittelpunkte von adsorbierten Molekülen des Lösungsmittels. Zur flüssigen Phase hin wird sie durch die äußere Helmholtz-Fläche (englisch outer Helmholtz plane, OHP) begrenzt und verläuft durch die Mittelpunkte der solvatisierten Ionen in ihrem Abstand der größten Annäherung an die Elektrode. Die diffuse Schicht liegt, wie im Stern-Modell, außerhalb der OHP. Die Ionen sind durch Moleküle des Lösungsmittels solvatisiert. Dadurch ergibt sich ein Aufbau aus drei Schichten: einer Lösungsmittelschicht (auch: innere Helmholtz-Schicht), einer Schicht aus solvatisierten Gegenionen (auch: äußere Helmholtz-Schicht) und einer diffusen Schicht. Außerdem beschrieb Grahame erstmals die Wirkung von Ionen, die ihre umhüllende Solvatationsschicht abgestreift haben und die Oberfläche der Elektroden berührten, obwohl eigentlich die Elektrodenoberfläche komplett mit solvatisierten Molekülen des Elektrolyt-Lösungsmittels bedeckt sein sollte. Die Anlagerung dieser Ionen an der metallischen Oberfläche einer Elektrode nannte er „spezifische Adsorption“.
Bockris-Müller-Devanathan-Modell
1963 formulierten dann der Elektrochemiker John O’Mara Bockris zusammen mit Klaus Müller und Michael Angelo Vincent Devanathan ihr Modell der unterschiedlichen Speicherprinzipien in elektrischen Doppelschichten, das zusätzlich zu den Vorstellungen der bisherigen Modelle auch noch den Einfluss des Lösungsmittels auf die Gesamtwirkung der Doppelschicht berücksichtigt. Mit diesem nach der Reihenfolge der Autorennamen in der Veröffentlichung genannten „BMD-Modell“ wurde mit der Beschreibung spezifisch adsorbierter Anionen auch die Redoxreaktion, die Grundlage der Pseudokapazität, genauer beschrieben.
Im Bild wird das BMD-Modell anschaulich dargestellt. An der geladenen Elektrode formen die an der Elektrodenoberfläche adsorbierten Lösungsmittelmoleküle die innere Helmholtz-Schicht. Die solvatisierten Kationen in der äußeren Helmholtz-Schicht, die sich direkt an die innere Helmholtz-Schicht anlagern, sind die Gegenionen zu den Ionen in der Elektrode und bilden die Doppelschichtkapazität. Dazwischen hat ein spezifisch adsorbiertes Kation die innere Helmholtz-Schicht durchdrungen, mit einer Redoxreaktion seine Ladung an die Elektrode abgegeben (Pseudokapazität) und ist dadurch zu einem Anion geworden.
Schmickler und Henderson
Wie sich experimentell gezeigt hat, hängen die Kapazitäten der inneren Doppelschicht auch deutlich vom verwendeten Elektrodenmetall ab. Die bisher erwähnten Modelle beschreiben alle die Verteilung der Ladungen im Elektrolyten und die daraus resultierende Doppelschichtkapazität, ohne auf die Eigenschaften des Elektrodenmaterials einzugehen. Sie können daher Unterschiede durch verschiedene Metalle nicht erklären. Ein erstes Doppelschichtmodell, das versucht hat, auch den Beitrag des Metalls wiederzugeben, wurde schon 1928 vorgestellt. Wie Schmickler und Henderson gezeigt haben, kann der Beitrag mancher Metalle zur Doppelschichtkapazität mit Hilfe des relativ einfachen Jellium-Modells abgeschätzt werden, das die Wechselwirkung des Elektronengases mit den Gitter der Metallionen beschreibt.
Trasatti-Buzzanca
Die weitere Forschung an Doppelschichten mit Elektroden aus Rutheniumdioxid führte 1971 durch Sergio Trasatti und Giovanni Buzzanca zur Erkenntnis, dass das elektrochemische Ladungsverhalten von spezifisch adsorbierten Ionen bei kleinen Spannungen dem von Kondensatoren gleicht. Die spezifisch adsorbierten Ionen lieferten einen Ladungstransfer zwischen dem Ion und der Elektrode und lieferten eine später sogenannte „Pseudokapazität“. Es war der erste Schritt in Richtung Pseudokondensatoren.
Conway
Zwischen 1975 und 1980 betrieb Brian Evans Conway Grundlagenforschung über Redox-Prozesse an mit Rutheniumoxid dotierten Elektroden. Er beschrieb 1991 den Übergang des Verhaltens eines Kondensators zu einer (wieder aufladbaren) Batterie (From Supercapacitor to Battery) bei der elektrochemischen Energiespeicherung und 1999 prägte er den Begriff „Superkondensator“ (englisch Supercapacitor) zur Kennzeichnung derjenigen Kondensatoren, die mit der faradayschen Ladungsspeicherung durch Redox-Reaktionen an den Elektroden-Oberflächen gegenüber der statischen Doppelschichtkapazität eine deutlich höhere Pseudokapazität aufweisen.
Die Kondensatorart, für die Conway den Begriff Superkondensator prägte, speicherte die elektrische Ladung überwiegend in Form der Pseudokapazität, ein Begriff, den Conway schon 1962 benutzte. Elektroden, die mit Metalloxiden oder leitfähigen Polymeren versehen waren, lieferten besonders hohe Werte an Pseudokapazität. Die Pseudokapazität, so konnte Conway feststellen, beruhte jedoch nicht nur auf „spezifisch adsorbierten Ionen“. Weitere Forschungsergebnisse lieferten im Wesentlichen drei Quellen für die Pseudokapazität: Redoxreaktionen, Interkalation und Elektrosorbtion. Letzteres ist eine unterpotentiale Deposition von Ad-Atomen.
Marcus-Theorie
Die physikalischen und mathematischen Grundlagen des Elektronen-Charge-Transfers ohne chemische Bindungen, der die Grundlage der Pseudokapazität ist, wurden beschrieben durch Rudolph Arthur Marcus. Die nach ihm benannte Marcus-Theorie beschreibt Redoxreaktionen (Einelektronenaustauschreaktionen), bei der das Lösungsmittel während der Reaktion bestimmend ist und erlaubt die Berechnung der Gibbs'schen freien Aktivierungsenthalpie aus den Polarisationseigenschaften des Lösungsmittels, der Größe und dem Abstand der Reaktanten bei der Elektronenübertragung und der freien Enthalpie der Redoxreaktion. Marcus erhielt für diese Theorie im Jahr 1992 den Nobelpreis für Chemie.
Anwendungen
Die Existenz einer elektrochemischen Doppelschicht, die biologische Makromolekülen (Proteine, Nukleinsäuren) in niedermolekularen Elektrolyten (Pufferlösungen) umgibt, ist wesentliche Voraussetzung für alle Methoden der Elektrophorese, die für die Biochemie der Makromoleküle von entscheidender Bedeutung wurde (siehe SDS-Gelelektrophorese, DNA-Sequenzierung). Gleiches gilt bei dem in der Praxis weniger bedeutsamen, oft störenden Phänomen der Elektroosmose. In der Elektrotechnik spielt das Phänomen der Doppelschicht in Superkondensatoren, auch „Doppelschichtkondensatoren“ genannt, als Summe der Doppelschichtkapazität und der Pseudokapazität eine wichtige Rolle.
Siehe auch
- Elektrochemie, für die die Doppelschicht zentral ist
- Debye-Hückel-Theorie, die analog zu den beschriebenen Modellen ist und ebenfalls die heute Debye-Länge genannte Größe benutzt
Literatur
- Volkmar M. Schmidt: Elektrochemische Verfahrenstechnik. Grundlagen, Reaktionstechnik, Prozessoptimierung. Wiley-VCH, Weinheim 2003, ISBN 3-527-29958-0.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 12.11. 2025