LCAO-Methode
Die LCAO-Methode (von engl. linear combination of atomic orbitals ‚lineare Kombination von Atomorbitalen‘) ist eine Quantensuperposition von Atomorbitalen und eine Methode zur Berechnung von Molekülorbitalen in der Quantenchemie. In der Quantenmechanik werden Elektronenkonfigurationen von Atomen als Wellenfunktion beschrieben, in Bezug auf Wasserstoff in der Schrödinger-Gleichung. In einer chemischen Reaktion werden die Orbitalwellenfunktionen modifiziert, d.h. die Elektronenwolke wird je nach den an einer chemischen Bindung teilnehmenden Atomen verändert.
Die LCAO-Methode wurde 1929 durch John Lennard-Jones mit der Beschreibung der Bindung diatomischer Moleküle der zweiten Periode veröffentlicht, wurde aber zuvor bereits durch Linus Pauling für H2+ verwendet.
Prinzip
Eine Grundannahme ist, dass die Anzahl der Molekülorbitale der Anzahl der Atomorbitale in der linearen Expansion gleicht. Die n Atomorbitale werden zu n Molekülorbitalen kombiniert, die mit einem Index von 1 bis n nummeriert werden und die nicht alle gleich sein müssen. Der Ausdruck der linearen Expansion für das i-te Molekülorbital ist:

oder

bei dem  (phi) ein Molekülorbital ist,
repräsentiert als Summe von n Atomorbitalen
(phi) ein Molekülorbital ist,
repräsentiert als Summe von n Atomorbitalen  (chi), jedes
multipliziert mit einem korrespondierenden Koeffizienten
(chi), jedes
multipliziert mit einem korrespondierenden Koeffizienten  ,
und r (nummeriert von 1 bis n) das im Term kombinierte Atomorbital
darstellt. Die Koeffizienten sind die Anteile des Beitrags der Atomorbitale am
Molekülorbital. Die Hartree-Fock-Methode
wird verwendet, um die Koeffizienten der Expansion zu ermitteln.
,
und r (nummeriert von 1 bis n) das im Term kombinierte Atomorbital
darstellt. Die Koeffizienten sind die Anteile des Beitrags der Atomorbitale am
Molekülorbital. Die Hartree-Fock-Methode
wird verwendet, um die Koeffizienten der Expansion zu ermitteln.
Die Orbitale werden als lineare Kombinationen der Basisfunktionen ausgedrückt, welche als Ein-Elektronen-Funktionen auf den Atomkernen der beteiligten Atome im Molekül zentriert sind. Die verwendeten Atomorbitale sind normalerweise vom Wasserstoff (z.B. Slater Type Orbitals), da diese analytisch bekannt sind, jedoch können auch andere ausgewählt werden, wie z.B. die Gauss-Orbitale im Standardbasissatz.
Durch die Minimierung der Gesamtenergie des Systems wird ein passender Satz an Koeffizienten der linearen Kombination bestimmt. Dieser quantitative Ansatz ist heute als Hartree-Fock-Methode bekannt. Seit der Einführung der molekularen Modellierung wird die LCAO-Methode jedoch weniger zur Optimierung einer Wellenfunktion als vielmehr zu einer qualitativen Bewertung herangezogen, welche bei der Vorhersage und bei der Erklärung der mit modernen Methoden erhaltenen Ergebnisse hilfreich ist. In diesem Fall werden die Form der Molekülorbitale und ihre jeweiligen Energien näherungsweise aus dem Vergleich der Energien der einzelnen Atomorbitale (oder Molekülfragmente) abgeleitet und die Level repulsion oder ähnliches angewendet. Die zur Klarstellung erzeugten Graphen werden als Korrelationsdiagramme (engl. correlation diagrams) bezeichnet. Die benötigten Energien der Atomorbitale stammen aus Berechnungen oder können über das Koopmans-Theorem experimentell bestimmt werden.
Der erste Schritt besteht aus der Zuweisung einer Punktgruppe zu dem Molekül. Ein häufiges Beispiel ist Wasser, welches eine C2v Symmetrie aufweist. Im Folgenden wird die reduzible Darstellung der Bindung im Wasser aufgeführt:
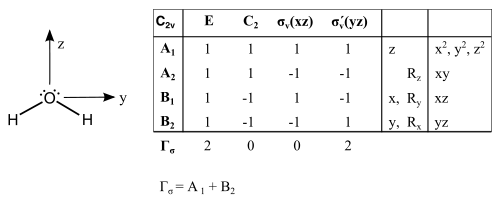
Jede Operation in der Punktgruppe wird in Bezug auf das Molekül durchgeführt. Die Anzahl an unveränderten Bindungen ist der Charakter einer Operation. Diese reduzible Darstellung wird in die Summe der irreduziblen Darstellungen zerlegt. Die irreduziblen Darstellungen korrespondieren mit der Symmetrie der beteiligten Orbitale.
MO-Diagramme bieten eine einfache qualitative Behandlung der LCAO-Näherung.
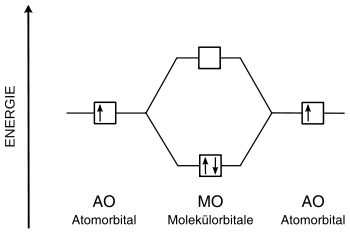
Quantitative Theorien sind die Hückel-Näherung, die erweiterte Hückel-Methode und die Pariser-Parr-Pople-Methode.
Betrachtet man ein System mit mehreren Elementen  (z.B. Atome), zentriert auf
(z.B. Atome), zentriert auf  ,
so stellt man fest, dass
,
so stellt man fest, dass  die Wellenfunktion des Elektrons beschreibt, wenn das Element
isoliert ist. Die Wellenfunktion
die Wellenfunktion des Elektrons beschreibt, wenn das Element
isoliert ist. Die Wellenfunktion  ,
die das Elektron im gesamten System beschreibt, kann durch eine lineare
Kombination von Wellenfunktionen
genähert werden:
,
die das Elektron im gesamten System beschreibt, kann durch eine lineare
Kombination von Wellenfunktionen
genähert werden:

Herleitung
Die Wellenfunktion
beschreibt ein Elektron, wenn das Element
isoliert ist.

Unter der Hypothese, dass die Größenordnung

nicht signifikativ ist außer für  ,
ist die potentielle Modifikation durch ein Element
,
ist die potentielle Modifikation durch ein Element  für die Wellenfunktion
für die Wellenfunktion  nicht so wichtig.
nicht so wichtig.
Jede Lösung der Gleichung des Gesamtsystems

kann durch eine lineare Kombination der isolierten Wellenfunktionen genähert werden:



© biancahoegel.de
Datum der letzten Änderung: Jena, den: 22.04. 2024