Geschwindigkeitsgesetz
Geschwindigkeitsgesetze oder Geschwindigkeitsgleichungen (englisch: rate laws) stellen in der chemischen Kinetik den Zusammenhang zwischen der Reaktionsgeschwindigkeit
 einerseits und Gehaltsgrößen von Ausgangsstoffen chemischer Reaktionen sowie konstanten Parametern andererseits
her.[1] Als Gehaltsgrößen werden üblicherweise Konzentrationen, teilweise jedoch auch andere
Maße für die Verfügbarkeit von Ausgangsstoffen, so etwa Partialdrücke oder relative Bedeckungen der Oberflächen von heterogenen Katalysatoren,[2][3] verwendet. Bei den konstanten Parametern kann es
sich um Geschwindigkeitskonstanten oder Halbwertszeiten sowie partielle Reaktionsordnungen
(siehe unten) der Ausgangsstoffe handeln.
einerseits und Gehaltsgrößen von Ausgangsstoffen chemischer Reaktionen sowie konstanten Parametern andererseits
her.[1] Als Gehaltsgrößen werden üblicherweise Konzentrationen, teilweise jedoch auch andere
Maße für die Verfügbarkeit von Ausgangsstoffen, so etwa Partialdrücke oder relative Bedeckungen der Oberflächen von heterogenen Katalysatoren,[2][3] verwendet. Bei den konstanten Parametern kann es
sich um Geschwindigkeitskonstanten oder Halbwertszeiten sowie partielle Reaktionsordnungen
(siehe unten) der Ausgangsstoffe handeln.
Geschwindigkeitsgesetze 0. bis 2. Ordnung
Allgemeine Formulierung
Häufig ist die Reaktionsgeschwindigkeit
einer chemischen Reaktion proportional zu Produkten von Potenzfunktionen der Konzentrationen der
Ausgangsstoffe. Sind an einer chemischen Reaktion
 Ausgangsstoffe
Ausgangsstoffe
 mit den Konzentrationen
mit den Konzentrationen
![{\displaystyle \mathrm {[Z_{j}]} }](/svg/9c2d16705f3fd3b15174cd6f96766a6b734da96b.svg) beteiligt, erhält man mit der Geschwindigkeitskonstante
beteiligt, erhält man mit der Geschwindigkeitskonstante
 Geschwindigkeitsgesetze der allgemeinen Form:
Geschwindigkeitsgesetze der allgemeinen Form:
![{\displaystyle r=k\prod _{j=1}^{N}[\mathrm {Z_{j}} ]^{z_{j}}}](/svg/e900ffddf44b6ef6b22df1c95a3faea1b3c400e1.svg)
Die Exponenten  bezeichnet man als partielle Reaktionsordnungen
in Bezug auf die Ausgangsstoffe
. Die Gesamtreaktionsordnung
bezeichnet man als partielle Reaktionsordnungen
in Bezug auf die Ausgangsstoffe
. Die Gesamtreaktionsordnung
 der betrachteten chemischen Reaktion ist gleich der Summe der partiellen Reaktionsordnungen
:
der betrachteten chemischen Reaktion ist gleich der Summe der partiellen Reaktionsordnungen
:

0. Ordnung
Geschwindigkeitsgesetze 0. Ordnung beschreiben chemische Reaktionen mit einer Gesamtreaktionsordnung
von null. Da dann üblicherweise auch die partiellen Reaktionsordnungen
in Bezug auf die Ausgangsstoffe
gleich null und die Potenzfunktionen
![{\displaystyle \mathrm {[Z_{j}]} ^{z_{j}}}](/svg/4b0f7d3626d6109bd480dee7ae1c6ebd322d6204.svg) der Konzentrationen der Ausgangsstoffe gleich eins werden,
wird die Reaktionsgeschwindigkeit
gleich der Geschwindigkeitskonstante
.
Geschwindigkeitsgesetze 0. Ordnung nehmen folglich folgende Form an:[4]
der Konzentrationen der Ausgangsstoffe gleich eins werden,
wird die Reaktionsgeschwindigkeit
gleich der Geschwindigkeitskonstante
.
Geschwindigkeitsgesetze 0. Ordnung nehmen folglich folgende Form an:[4]
![{\displaystyle r=k\cdot [\mathrm {A} ]^{0}=k}](/svg/7aae6990e2bca13001aa5bf10a0d486eca0e7d16.svg)
Die Reaktionsgeschwindigkeit ist somit unabhängig von den Konzentrationen
der Ausgangsstoffe. Geschwindigkeitsgesetze 0. Ordnung
können beispielsweise bei heterogenen Reaktionen auftreten.[3][4][5] Weiterhin weisen viele enzymatische Katalysen eine Kinetik 0. Ordnung auf,
sofern das Substrat in großem Überschuss vorliegt.[5][6]
1. Ordnung
Geschwindigkeitsgesetze 1. Ordnung beschreiben chemische Reaktionen mit einer Gesamtreaktionsordnung
von eins. Üblicherweise ist die partielle Reaktionsordnung
 in Bezug auf einen Ausgangsstoff
in Bezug auf einen Ausgangsstoff
 gleich eins, während alle anderen partiellen Reaktionsordnungen
in Bezug auf eventuell vorhandene weitere Ausgangsstoffe
gleich null sind. Entsprechend werden die Potenzfunktionen
der Konzentrationen eventuell vorhandener weiterer Ausgangsstoffe
gleich eins, und die Potenzfunktion
gleich eins, während alle anderen partiellen Reaktionsordnungen
in Bezug auf eventuell vorhandene weitere Ausgangsstoffe
gleich null sind. Entsprechend werden die Potenzfunktionen
der Konzentrationen eventuell vorhandener weiterer Ausgangsstoffe
gleich eins, und die Potenzfunktion
![{\displaystyle \mathrm {[A]} ^{a}}](/svg/3a5882c3c5035cb05a00b72ff28a78743e4bbe67.svg) der Konzentration des Ausgangsstoffes
gleich der Konzentration
der Konzentration des Ausgangsstoffes
gleich der Konzentration
![{\displaystyle {\ce {[A]}}}](/svg/881146b6653b24508d87e34a81c84832f1d5ffea.svg) . Dann hängt die Reaktionsgeschwindigkeit
vom Produkt der Geschwindigkeitskonstante
mit
ab, so dass Geschwindigkeitsgesetze 1. Ordnung folgende Form annehmen:
. Dann hängt die Reaktionsgeschwindigkeit
vom Produkt der Geschwindigkeitskonstante
mit
ab, so dass Geschwindigkeitsgesetze 1. Ordnung folgende Form annehmen:
![{\displaystyle r=k\cdot [\mathrm {A} ]}](/svg/93fcd4f20d5f57a82a50b8c61df3528aca86a861.svg)
Typischerweise sind unimolekulare Zerfallsreaktionen mit einer Molekularität von eins Reaktionen 1. Ordnung. Ein Beispiel hierfür sind radioaktive Zerfallsreaktionen. Ebenfalls erster Ordnung sind chemische Reaktionen, die als langsamste Elementarreaktion und damit als geschwindigkeitsbestimmenden Schritt einen unimolekularen Zerfall umfassen. Beispiele hierfür sind Eliminierungen und nukleophile Substitutionen 1. Ordnung.
2. Ordnung
Geschwindigkeitsgesetze 2. Ordnung beschreiben die Kinetik von Elementarreaktionen mit einer Molekularität von zwei, die einen bimolekularen Stoß umfassen. Umfasst die
betrachtete Reaktion einen Stoß zweier gleichartiger Teilchen
, wird die Reaktionsgleichung:

Die Reaktionsgeschwindigkeit ist dann proportional zum Quadrat
![{\displaystyle {\ce {[A]^2}}}](/svg/32c53590d86d69973f7b348ed51ed59d2fd06e94.svg) der Konzentration
des Ausgangsstoffes
. Die partielle Reaktionsordnung in Bezug auf
ist gleich der Gesamtreaktionsordnung und damit gleich zwei.
Das Geschwindigkeitsgesetz wird:
der Konzentration
des Ausgangsstoffes
. Die partielle Reaktionsordnung in Bezug auf
ist gleich der Gesamtreaktionsordnung und damit gleich zwei.
Das Geschwindigkeitsgesetz wird:
![{\displaystyle r=k\cdot [\mathrm {A} ]^{2}}](/svg/bf3e440bdd3772391f76207d46965e8020415853.svg)
Ein derartiges Szenario kann beispielsweise bei Reaktionen auftreten, die nach dem Lindemann-Mechanismus ablaufen.
Weiterhin beschreiben Geschwindigkeitsgesetze 2. Ordnung die Kinetik von Elementarreaktionen mit einer Molekularität von zwei, die einen bimolekularen Stoß zweier verschiedenartiger Teilchen
und
 umfassen. Die Reaktionsgleichung lautet dann:
umfassen. Die Reaktionsgleichung lautet dann:
Die Reaktionsgeschwindigkeit ist dann proportional zum Produkt der Konzentrationen
und
![{\displaystyle {\ce {[B]}}}](/svg/43c539b685a181af4359a0c94b23d232761c3340.svg) der Ausgangsstoffe
und
.
Die partiellen Reaktionsordnungen in Bezug auf die Ausgangsstoffe
und
sind jeweils eins. Das Geschwindigkeitsgesetz wird entsprechend:
der Ausgangsstoffe
und
.
Die partiellen Reaktionsordnungen in Bezug auf die Ausgangsstoffe
und
sind jeweils eins. Das Geschwindigkeitsgesetz wird entsprechend:
![{\displaystyle r=k\cdot [\mathrm {A} ]\cdot [\mathrm {B} ]}](/svg/0d75a6af00e5a718a044cad7ff42f9163512cd28.svg)
Beispiele hierfür sind bimolekulare konzertierte Elementarreaktionen, wie Eliminierungen und nukleophile Substitutionen 2. Ordnung.
Pseudo-Reaktionsordnungen
Betrachtet wird beispielhaft eine Elementarreaktion 2. Ordnung, an der Ausgangsstoff
mit der Konzentration
sowie Ausgangsstoff
mit der Konzentration
teilnehmen.
Wenn die partiellen Reaktionsordnungen in Bezug auf
und
jeweils eins sind, lautet das Geschwindigkeitsgesetz:
![{\displaystyle r=k[{\ce {A}}][{\ce {B}}]}](/svg/cfd0d3bae7ba5c56f4e78b68bf3881d2c96c5230.svg)
Die Elementarreaktion kann nun eine Pseudo-Ordnung aufweisen, die den Reaktionsmechanismus und die Molekularität nicht widerspiegelt, weil
im Verlauf der Reaktion (näherungsweise) konstant bleibt.
Dies kann der Fall sein, wenn es sich bei
um einen regenerierenden
Katalysator handelt, wenn
in einem offenen Reaktionssystem kontinuierlich nachgeliefert wird oder wenn
![{\displaystyle {\ce {[B] >> [A]}}}](/svg/74fe070a0634f4c32a066662bffa296ebe933913.svg) ist.
In derartigen Fällen können die Geschwindigkeitskonstante
und
zu einer Pseudo-Konstante
ist.
In derartigen Fällen können die Geschwindigkeitskonstante
und
zu einer Pseudo-Konstante
 zusammengefasst werden:
zusammengefasst werden:
![{\displaystyle k'=k[{\ce {B}}]}](/svg/c2549946acd88fd36b67e3d2fa8cf0fcc73bad0c.svg)
Damit vereinfacht sich das Geschwindigkeitsgesetz 2. Ordnung zu einem Geschwindigkeitsgesetz, in dem die partielle Reaktionsordnung von
, die gleich eins ist, anscheinend zur Gesamtreaktionsordnung wird:
![{\displaystyle r=k[{\ce {A}}][{\ce {B}}]=k'[{\ce {A}}]}](/svg/6dad53740f8638e5eb18169797d38849fb5bcc1b.svg)
Anscheinende Gesamtreaktionsordnungen von Elementarreaktionen, die sich von der Molekularität letzterer unterscheiden, bezeichnet man als Pseudo-Reaktionsordnungen.[7]
Im obigen Beispiel ist das Geschwindigkeitsgesetz
![{\displaystyle r=k'[{\ce {A}}]}](/svg/aa300f267f698ad37a98809f7f248b3f7d2022c9.svg) pseudo-erster Ordnung.
pseudo-erster Ordnung.
Ein Beispiel für eine Reaktion pseudo-erster Ordnung ist Hydrolyse von Estern in wässeriger Lösung:

Die partiellen Reaktionsordnungen in Bezug auf
 und
und
 sind jeweils eins, die Gesamtreaktionsordnung ist zwei.
Das Geschwindigkeitsgesetz der Reaktion lautet entsprechend:
sind jeweils eins, die Gesamtreaktionsordnung ist zwei.
Das Geschwindigkeitsgesetz der Reaktion lautet entsprechend:
![{\displaystyle {\ce {{\it {r}}\;=\;{\it {k}}\cdot [CH3COOCH3]\cdot [H2O]}}}](/svg/0115ee85742846023f6d6dc41fd86ed7d17debec.svg)
Da jedoch der Ausgangsstoff Wasser gleichzeitig als Lösungsmittel fungiert, gilt:
![{\displaystyle {\ce {[CH3COOCH3] << [H2O]}}}](/svg/895ed358dc6560b42f5e118ac2d040416700875c.svg)
Somit kann ![{\displaystyle {\ce {[H2O]}}}](/svg/938697d1349a832a18be1d02c3e5202ea8d5ec47.svg) als konstant angesehen werden. Mit
als konstant angesehen werden. Mit
![{\displaystyle {\ce {{\it {k}}'\;=\;{\it {k}}\cdot [H2O]}}}](/svg/d1033c19f4dfa158f0cf87fbad6b3f8499fbdbe7.svg) erhält man ein Geschwindigkeitsgesetz mit der Pseudo-Reaktionsordnung eins:
erhält man ein Geschwindigkeitsgesetz mit der Pseudo-Reaktionsordnung eins:
![{\displaystyle {\ce {{\it {r}}\;=\;{\it {k}}'\cdot [CH3COOCH3]}}}](/svg/e8486229834449a863764ae7e4b30a7ff22471c9.svg)
Ebenso ist die Rohrzuckerinversion, die Reaktion von Saccharose (Rohrzucker) und Wasser zu D-Glucose und D-Fructose (Invertzucker), eigentlich eine Reaktion 2. Ordnung, wobei die partiellen Reaktionsordnungen in Bezug auf die Ausgangsstoffe jeweils eins sind:

Da das Lösungsmittel Wasser in großem Überschuss vorliegt, findet man auch für diese Reaktion die pseudo-Reaktionsordnung eins.[7]
Bestimmung partieller Reaktionsordnungen
Integrationsmethode
Zeitgesetze geben die Konzentration }](/svg/ae3cb4aa5453bac2631e10c730a89df05f76c7cd.svg) eines an einer Reaktion beteiligten Eduktes
als Funktion der Reaktionsdauer
eines an einer Reaktion beteiligten Eduktes
als Funktion der Reaktionsdauer
 an. In einigen Fällen lassen sich Zeitgesetze in Form von Geradengleichungen angeben
(siehe Kinetik (Chemie)). Ist die partielle Reaktionsordnung in Bezug auf
gleich 0, muss die Auftragung von
an. In einigen Fällen lassen sich Zeitgesetze in Form von Geradengleichungen angeben
(siehe Kinetik (Chemie)). Ist die partielle Reaktionsordnung in Bezug auf
gleich 0, muss die Auftragung von
}](/svg/9e47769cea081ce61756fd186559d7558227a776.svg) gegen
eine Gerade ergeben. Ist die partielle Reaktionsordnung in Bezug auf
gleich 1, muss die Auftragung von
gegen
eine Gerade ergeben. Ist die partielle Reaktionsordnung in Bezug auf
gleich 1, muss die Auftragung von
/[{\text{A}}]_{0}\right)}](/svg/bdf79434d4a77fc19c031ad735f81b863c3a33cb.svg) gegen
eine Gerade ergeben. Ist die partielle Reaktionsordnung in Bezug auf
gleich 2, muss die Auftragung von
gegen
eine Gerade ergeben. Ist die partielle Reaktionsordnung in Bezug auf
gleich 2, muss die Auftragung von
}](/svg/e124fad820744838f97027a4f29e81c9e3b9dac8.svg) gegen
eine Gerade ergeben.
Sofern aus einer dieser Auftragungen mit experimentell erhaltenen Wertepaaren eine Regressionsgerade mit einem zufriedenstellenden
Bestimmtheitsmaß resultiert, spricht dies dafür, dass die partielle Reaktionsordnung von
den entsprechenden Wert besitzt.[8][9][10]
gegen
eine Gerade ergeben.
Sofern aus einer dieser Auftragungen mit experimentell erhaltenen Wertepaaren eine Regressionsgerade mit einem zufriedenstellenden
Bestimmtheitsmaß resultiert, spricht dies dafür, dass die partielle Reaktionsordnung von
den entsprechenden Wert besitzt.[8][9][10]
Methode der Anfangsgeschwindigkeit
Der natürliche Logarithmus eines Geschwindigkeitsgesetzes, in dem die Reaktionsgeschwindigkeit gleich einem Produkt von Potenzfunktionen der Konzentrationen der Edukte gesetzt wird, ist (da Einheiten nicht logarithmierbar sind, würde man vor dem Logarithmieren beide Seiten des Geschwindigkeitsgesetzes formal durch die Einheit der Reaktionsgeschwindigkeit teilen):
![{\displaystyle \ln r=\ln k+\sum \limits _{j=1}^{N}\ln[{\ce {Z}}_{j}]^{z_{j}}=\ln k+\sum \limits _{j=1}^{N}(z_{j}\cdot \ln[{\ce {Z}}_{j}])}](/svg/790b2166cddafd29342b4f47431ec43c3231c9f2.svg)
Im Falle einer Reaktion 2. Ordnung erhält man mit
und
 als den partiellen Reaktionsordnungen der Edukte
und
entsprechend:
als den partiellen Reaktionsordnungen der Edukte
und
entsprechend:
![{\displaystyle \ln r=\ln k+a\ln[{\ce {A}}]+b\ln[{\ce {B}}]}](/svg/32ee26f938d8d1c3a487e6e7abfaab1f591d9ddd.svg)
Zur Abschätzung der partiellen Reaktionsordnung
von Ausgangsstoff
führt man mehrere Versuchsansätze mit verschiedenen Anfangskonzentrationen
![{\displaystyle {\ce {[A]_0}}}](/svg/9697f40eac0e29cf11c9e33aed6ea5e5f6118ad9.svg) von
durch. Die Anfangskonzentration
von
durch. Die Anfangskonzentration
![{\displaystyle {\ce {[B]_0}}}](/svg/36737c191269a27ab640fcc009bb44653f17215c.svg) von Ausgangsstoff
hält man hingegen konstant.
Man misst dann die Anfangsreaktionsgeschwindigkeit
von Ausgangsstoff
hält man hingegen konstant.
Man misst dann die Anfangsreaktionsgeschwindigkeit
 unmittelbar nach Start der Reaktion. Für kleine Reaktionsdauern
gilt näherungsweise
unmittelbar nach Start der Reaktion. Für kleine Reaktionsdauern
gilt näherungsweise
\approx [\mathrm {A} ]_{0}}](/svg/57652bf9b22f46f87b66cb0bec6a5817bcd103e5.svg)
sowie
\approx [\mathrm {B} ]_{0}={\text{Konstante}}}](/svg/2ad678330d493027952a0ec7bab81055c205c2e6.svg) .
.
Dann erhält man:
![{\displaystyle \ln r_{0}\approx a\ln[{\ce {A}}]_{0}+{\textrm {Konstante}}}](/svg/e6b94e8cb405c0b5e9889d3f9d000ce53277640e.svg)
Der Logarithmus der Anfangsgeschwindigkeit  als Funktion von
als Funktion von
![{\displaystyle {\ce {ln [A]_0}}}](/svg/957548cce011bece64ef24b1cba60208eab6709f.svg) sollte dann eine Gerade ergeben, deren
Steigung
ist. Ein Nachteil dieser Methode ist, dass zur Bestimmung von
sollte dann eine Gerade ergeben, deren
Steigung
ist. Ein Nachteil dieser Methode ist, dass zur Bestimmung von
 relativ kleine Änderungen von
gemessen werden müssen, so dass erhebliche Anstrengungen in die
Minimierung von Messfehlern investiert werden müssen.[10][11]
relativ kleine Änderungen von
gemessen werden müssen, so dass erhebliche Anstrengungen in die
Minimierung von Messfehlern investiert werden müssen.[10][11]
Halbwertszeitverfahren
Partielle Reaktionsordnungen in Bezug auf einen Ausgangsstoff
lassen sich mittels der Halbwertszeiten
 ermitteln,
also derjenigen Reaktionsdauer, innerhalb derer die Anfangskonzentration
von
auf ihren halben Wert
ermitteln,
also derjenigen Reaktionsdauer, innerhalb derer die Anfangskonzentration
von
auf ihren halben Wert
=1/2\cdot [\mathrm {A} ]_{0}}](/svg/d6d0c1c37a5a4b16c4156fa582df7337a6e7ddd3.svg) gefallen ist.
Der stöchiometrische Koeffizient des Ausgangsstoffes
sei, konventionsgemäß mit negativem Vorzeichen,
gefallen ist.
Der stöchiometrische Koeffizient des Ausgangsstoffes
sei, konventionsgemäß mit negativem Vorzeichen,
 . Zur Bestimmung der partiellen Reaktionsordnung von
wird
für verschiedene Werte von
ermittelt.[8][12]
. Zur Bestimmung der partiellen Reaktionsordnung von
wird
für verschiedene Werte von
ermittelt.[8][12]
Ist die partielle Reaktionsordnung von
gleich 0, wird:
![{\displaystyle t_{1/2}=-{\frac {[{\text{A}}]_{0}}{2\nu _{\text{A}}k}}}](/svg/8e03ed1833f5c5375546c8ae24abbab3cf6cc86f.svg)
Es muss dann also gelten: ![{\displaystyle t_{1/2}\sim [\mathrm {A} ]_{0}}](/svg/e0e5dbb49769fd777e4552bbe38ff9f8f264cb23.svg)
Ist die partielle Reaktionsordnung von
gleich 1, gilt:

Dann ist unabhängig von
![{\displaystyle [\mathrm {A} ]_{0}}](/svg/500af2b45c7d2bf925a8be44a98c3e2b55588b8e.svg) .
.
Ist die partielle Reaktionsordnung von
gleich 2, gilt:
![{\displaystyle t_{1/2}=-{\frac {1}{\nu _{\text{A}}k[{\text{A}}]_{0}}}}](/svg/c46c38104c9b3086775761f1e6b6faaf32d58f24.svg)
Logarithmieren ergibt:
![{\displaystyle \ln t_{1/2}=\ln \left(-{\frac {1}{\nu _{\text{A}}k}}\right)+\ln {\frac {1}{[{\text{A}}]_{0}}}}](/svg/afe51e9891f13fd6582abe919572affdf3b38c27.svg)
Es muss dann also gelten:
![{\displaystyle \ln t_{1/2}\sim \ln {\frac {1}{[{\text{A}}]_{0}}}}](/svg/eda924eabc269f9cec12534c77317cd304d559b2.svg)
Überschuss-Methode
Nach Wilhelm Ostwald kann die partielle Reaktionsordnung
abgeschätzt werden, wenn alle anderen Ausgangsstoffe in so großem Überschuss vorliegen, dass deren Konzentration als konstant betrachtet werden kann. Dies kann beispielsweise der Fall sein, wenn
ein gelöster Stoff ist und ein zweiter Ausgangsstoff
gleichzeitig als
Lösungsmittel fungiert. Die Konzentration
von
wird für verschiedene Reaktionsdauern
gemessen. Für eine Reaktion
 wird das Geschwindigkeitsgesetz mit
und
als partiellen Reaktionsordnungen von
und
:[13]
wird das Geschwindigkeitsgesetz mit
und
als partiellen Reaktionsordnungen von
und
:[13]
![{\displaystyle r=k\cdot [{\rm {A}}]^{a}\cdot [{\rm {B}}]^{b}}](/svg/ecc1a6d4bb8e4712c56052e684d88cad8ab2f179.svg)
Da und folglich
![{\displaystyle {\ce {[B]^{b}}}}](/svg/fac656070a83aebb619ec13516e564a4060e241c.svg) als konstant betrachtet werden können, wird mit
als konstant betrachtet werden können, wird mit
![{\displaystyle k'=k\cdot [{\rm {B}}]^{b}}](/svg/7ba44812f607ed1a3b2e018a1af5a10b96b09197.svg) :
:
![{\displaystyle r=k'\cdot [{\rm {A}}]^{a}}](/svg/cdf96648cd7ec7f69a4ec3c54dda5fb599d02542.svg)
Dieser Ausdruck wird logarithmiert. Die partielle Reaktionsordnung
von
lässt sich als Steigung der so erhaltenen Gerade ermitteln:
![{\displaystyle \ln r=\ln k'+a\cdot \ln[{\rm {A}}]}](/svg/1c1608e1b9893aba7f4d5d7ffdc7f4a07565b8a0.svg)
Komplexe Geschwindigkeitsgesetze
Hintergrund
Viele chemische Bruttoreaktionen sind durch komplexe Reaktionsmechanismen gekennzeichnet, die eine Reihe von konsekutiv oder parallel ablaufenden Elementarreaktionen
umfassen, die in die Bruttogeschwindigkeitsgesetze der Gesamtreaktionen eingehen. Sofern die Rückreaktionen von Elementarreaktionen nennenswerte Geschwindigkeiten aufweisen, müssen diese bei der Aufstellung von Bruttogeschwindigkeitsgesetzen für
Gesamtreaktionen berücksichtigt werden. Weiterhin können Szenarios existieren, in denen Konkurrenzreaktionen in die Geschwindigkeitsgesetze der Reaktionen von Interesse eingehen. Hieraus resultieren oft komplexe Geschwindigkeitsgesetze,
die Brüche und/oder mehrere Geschwindigkeitskonstanten und/oder gemischte Reaktionsordnungen in Bezug auf bestimmte Ausgangsstoffe und/oder gebrochene Reaktionsordnungen in Bezug auf bestimmte Ausgangsstoffe enthalten. Teilweise lassen
sich Geschwindigkeitsgesetze durch Näherungen vereinfachen. Beispielsweise lässt sich die Kinetik von zusammengesetzten Reaktionen, in deren Verlauf mehrere konsekutive Elementarreaktionen auftreten, häufig in zufriedenstellender Weise durch
die Kinetik einer langsam ablaufenden Elementarreaktion, die als geschwindigkeitsbestimmender Schritt bezeichnet wird, annähern. Treten im Verlauf von zusammengesetzten Reaktionen reaktive Intermediate auf,
kann die Beschreibung der Reaktionskinetik durch das Bodensteinsche Quasistationaritätsprinzip vereinfacht werden.
Andererseits kann aus Geschwindigkeitsgesetzen nicht unmittelbar auf den Reaktionsmechanismus geschlossen werden.[14] Ein Beispiel ist die Reaktion von Iod  und
Wasserstoff
und
Wasserstoff  zu Iodwasserstoff
zu Iodwasserstoff
 :
:

Die beobachtete phänomenologische Reaktionskinetik ergibt anscheinende partielle Reaktionsordnungen von eins in Bezug auf
und
sowie eine anscheinende Gesamtreaktionsordnung von zwei. Dies ist mit der Vorstellung kompatibel, dass eine einen bimolekularen Stoß von
mit
umfassende bimolekulare Elementarreaktion stattfindet. Tatsächlich ist dies nicht der Fall; die zu
führenden Reaktionspfade beinhalten vielmehr die Reaktion von Iod-Radikalen
 .[15][16]
.[15][16]
Gebrochene Reaktionsordnungen
Zusammengesetzte und gekoppelte Reaktionen weisen häufig gebrochene Reaktionsordnungen wie 1,5 auf, die keine natürlichen Zahlen sind. Ein Beispiel hierfür sind radikalische Polymerisationen, in deren Verlauf ein Initiator
 und ein Monomer
und ein Monomer
 zu einem Polymer
zu einem Polymer
 reagieren. Zunächst zerfällt
in Radikale
reagieren. Zunächst zerfällt
in Radikale
 :
:

Hierauf folgt der Kettenstart:

Auf den Kettenstart folgt das Kettenwachstum:

Zu berücksichtigen sind außerdem die Kettenabbruchreaktionen. Kettenabbruch kann einerseits durch Rekombination wachsender Kettenenden stattfinden:

Andererseits kann Kettenabbruch durch Disproportionierung erfolgen:

Betrachtet man die Konzentration der Kettenträger
 unter Anwendung des Bodensteinschen Quasistationaritätsprinzips näherungsweise als konstant, weisen radikalische Polymerisation bei großen Polymerisationsgraden
eine Gesamtreaktionsordnung von 1,5 auf. Die partielle Reaktionsordnung in Bezug auf Monomer
ist 1, die partielle Reaktionsordnung in Bezug auf den Initiator
0,5. Mit der Geschwindigkeitskonstanten
erhält man so als Geschwindigkeitsgesetz:[17]
unter Anwendung des Bodensteinschen Quasistationaritätsprinzips näherungsweise als konstant, weisen radikalische Polymerisation bei großen Polymerisationsgraden
eine Gesamtreaktionsordnung von 1,5 auf. Die partielle Reaktionsordnung in Bezug auf Monomer
ist 1, die partielle Reaktionsordnung in Bezug auf den Initiator
0,5. Mit der Geschwindigkeitskonstanten
erhält man so als Geschwindigkeitsgesetz:[17]
![{\displaystyle r=k\cdot [{\rm {M]\cdot [{\rm {I]^{1/2}}}}}}](/svg/8e8dfa91a530d63c039691b767a30d161e67546c.svg)
Gemischte Reaktionsordnungen
In komplexen Geschwindigkeitsgesetzen kann ein Ausgangsstoff
mit verschiedenen partiellen Reaktionsordnungen auftreten. Derartige Geschwindigkeitsgesetze enthalten mindestens zwei Potenzfunktionen der Konzentration
von
, die nicht zu einer Potenzfunktion zusammengefasst
werden können. Ein Beispiel hierfür ist:
![{\displaystyle r=k_{1}[A]+k_{2}[A]^{2}}](/svg/d90ff7734705f2886b4c6fe5f76b1915511cc6ed.svg)
Hierbei tritt im ersten
Summand mit der partiellen Reaktionsordnung (oder der pseudo-Reaktionsodnung) eins, im zweiten Summand mit der partiellen Reaktionsordnung (oder der pseudo-Reaktionsordnung) zwei auf.
Man spricht dann von gemischter 1. und 2. Reaktionordnung.[18] Sofern die Geschwindigkeitskonstanten
 und
und
 vergleichbare Werte haben, weist die Reaktion bei kleinen Werten von
näherungsweise eine Kinetik 1. Ordnung (oder pseudo-erster Ordnung) auf, bei großen Werten von
eine Kinetik 2. Ordnung (oder pseudo-zweiter Ordnung). Daher kann eine erartige Reaktion zunächst eine phänomenologische Reaktionsordnung von zwei aufweisen, die aufgrund des Verbrauchs des Ausgangsstoffes
abnimmt, bis die phänomenologische beobachtbare Reaktionsordnung eins wird.
vergleichbare Werte haben, weist die Reaktion bei kleinen Werten von
näherungsweise eine Kinetik 1. Ordnung (oder pseudo-erster Ordnung) auf, bei großen Werten von
eine Kinetik 2. Ordnung (oder pseudo-zweiter Ordnung). Daher kann eine erartige Reaktion zunächst eine phänomenologische Reaktionsordnung von zwei aufweisen, die aufgrund des Verbrauchs des Ausgangsstoffes
abnimmt, bis die phänomenologische beobachtbare Reaktionsordnung eins wird.
Geschwindigkeitsgesetze können weiterhin Brüche mit zwei oder mehreren Termen enthalten. Ein Grund hierfür ist, dass für Reaktionen mit komplexen Mechanismen für verschiedene Konzentrationen der Ausgangsstoffe verschiedene Elementarreaktionen
als langsamster und damit geschwindigkeitsbestimmender Schritt fungieren. Ein Beispiel ist die Oxidation von Alkoholen zu
Ketonen durch Hexacyanoferrat(III)-Ionen
![{\displaystyle {\ce {[Fe(CN)6]^{3-}}}}](/svg/f48cbd9613eea35d3359a8be472ebabc2b622d53.svg) unter Katalyse mit Ruthenium(VI)-Ionen
unter Katalyse mit Ruthenium(VI)-Ionen
 .[19]
Das Geschwindigkeitsgesetz lautet:
.[19]
Das Geschwindigkeitsgesetz lautet:
![{\displaystyle r={\frac {\ce {{\big [}[Fe(CN)6]^{3-}{\big ]}}}{k_{\alpha }+k_{\beta }{\ce {{\big [}[Fe(CN)6]^{3-}{\big ]}}}}}}](/svg/025bd3cc1df58a0dbb6dfd2315ea67e2fb90979c.svg)
Hierbei sind  und
und
 Geschwindigkeitskonstanten. Wenn
Geschwindigkeitskonstanten. Wenn
![{\displaystyle {\ce {{\it {k}}_{\alpha }<<{\it {k}}_{\beta }{\big [}[Fe(CN)6]^{3-}{\big ]}}}}](/svg/9a37d37261de6ff24b796b94b099b6d9fd617a07.svg) ist, wäre die partielle Reakionsordnung in Bezug auf
sowie die Gesamtreaktionsordnung null, und die Pseudogeschwindigkeitskonstante der Reaktion wäre
ist, wäre die partielle Reakionsordnung in Bezug auf
sowie die Gesamtreaktionsordnung null, und die Pseudogeschwindigkeitskonstante der Reaktion wäre
 .
Wird durch den sukzessiven Verbrauch von
hingegen
.
Wird durch den sukzessiven Verbrauch von
hingegen
![{\displaystyle {\ce {{\it {k}}_{\alpha }>>{\it {k}}_{\beta }{\big [}[Fe(CN)6]^{3-}{\big ]}}}}](/svg/a75fd074fabed94936cd802d0ea04f06ece50b18.svg) ,
wird die partielle Reaktionsordnung in Bezug auf
sowie die gesamte Reaktionsordnung eins, und die Pseudogeschwindigkeitskonstante der Reaktion wäre
,
wird die partielle Reaktionsordnung in Bezug auf
sowie die gesamte Reaktionsordnung eins, und die Pseudogeschwindigkeitskonstante der Reaktion wäre
 .
.
Weitere Beispiele für Reaktionsmechanismen, die zu Geschwindigkeitsgesetzen führen, in denen Zähler mit zwei Termen mit verschiedenen Reaktionsordnungen in Bezug auf einen Ausgangsstoff auftreten, sind der Michaelis–Menten-Mechanismus für Enzymkatalyse und der Lindemann-Mechanismus.
Beispiele
Wasserstoffoxidation
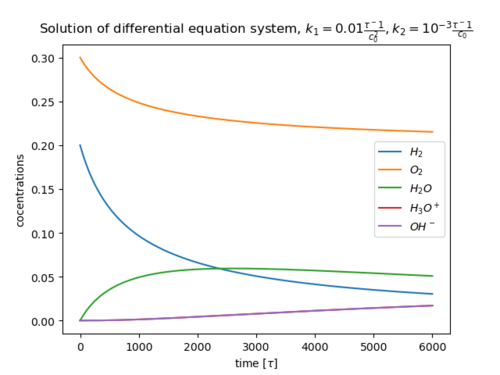
 und die Anfangskonzentrationen sind in diesem Beispiel beliebig gewählt.
und die Anfangskonzentrationen sind in diesem Beispiel beliebig gewählt.
 ist eine Referenzkonzentration.
ist eine Referenzkonzentration.Als Beispiel für ein System aus Differentialgleichungen, die jeweils Geschwindigkeitsgesetze von Elementarreaktionen sind und die für die Kinetik einer Gesamtreaktion relevant sind, wird die Oxidation von Wasserstoff herangezogen:

|
(Geschwindigkeitskonstante:
 ) )
|
ein Teil dissoziiert

|
(Geschwindigkeitskonstante:
 ) )
|
Die Geschwindigkeitsgesetze (Gl.1) für die fünf Spezies lauten:
![{\displaystyle {\frac {\mathrm {d} }{\mathrm {d} t}}\mathrm {[H_{2}]} =-2k_{1}\mathrm {[H_{2}]^{2}[O_{2}]} }](/svg/2c13665e60d079c9c00548aa0321ec4bab0f19bf.svg)
![{\displaystyle {\frac {\mathrm {d} }{\mathrm {d} t}}\mathrm {[O_{2}]} =-k_{1}\mathrm {[H_{2}]^{2}[O_{2}]} }](/svg/b250606928c4804538db7c25ae171f3814b10de8.svg)
![{\displaystyle {\frac {\mathrm {d} }{\mathrm {d} t}}\mathrm {[H_{2}O]} =+2k_{1}\mathrm {[H_{2}]^{2}[O_{2}]} -2k_{2}\mathrm {[H_{2}O]^{2}} }](/svg/2ff8e0597eaea466b1d3a2394ae0d6e191450de3.svg)
![{\displaystyle {\frac {\mathrm {d} }{\mathrm {d} t}}\mathrm {[H_{3}O^{+}]} =+k_{2}\mathrm {[H_{2}O]^{2}} }](/svg/008993d5ea22cfbbff2fadbb68eb25d19621909a.svg)
![{\displaystyle {\frac {\mathrm {d} }{\mathrm {d} t}}\mathrm {[OH^{-}]} =+k_{2}\mathrm {[H_{2}O]^{2}} }](/svg/56a36873b3b7c5d6c84f6c7fb5058e45703f27e4.svg)
Einzelnachweise
- ↑ Eintrag zu rate law (empirical differential rate equation). In: IUPAC
(Hrsg.): Compendium of Chemical Terminology. The “Gold Book”.
doi:
 10.1351/goldbook.R05141 – Version: 5.0.0.
10.1351/goldbook.R05141 – Version: 5.0.0.
- ↑ Wladimir Reschetilowski: Kinetik heterogen katalysierter Reaktionen und Reaktionsmechanismen. In:
Einführung in die Heterogene Katalyse. Springer Berlin Heidelberg, Berlin, Heidelberg 2015, ISBN 978-3-662-46983-5, "7.1.1 Oberflächenreaktion als geschwindigkeitsbestimmender Schritt" und "7.1.2 Kinetische
Ansätze nach Hougen-Watson", S. 100–121,
doi: 10.1007/978-3-662-46984-2_7.
- ↑ Hochspringen nach: a b Carl Heinz Hamann, Dirk Hoogestraat, Rainer Koch: Grundlagen der Kinetik:
Von Transportprozessen zur Reaktionskinetik. Springer, Berlin, Heidelberg 2017, ISBN 978-3-662-49392-2, 4.2.3.3 Heterogene Katalyse, S. 152 ff.,
doi: 10.1007/978-3-662-49393-9.
- ↑ Hochspringen nach: a b Gerd Wedler, Hans-Joachim Freund: Lehr- und Arbeitsbuch Physikalische Chemie. 7. Auflage. Wiley-VCH, Weinheim 2019, ISBN 978-3-527-82727-5, 1.5.5 Reaktionen nullter Ordnung.
- ↑ Hochspringen nach: a b Peter W. Atkins, Julio de Paula: Physikalische Chemie. 5. Auflage. Wiley-VCH, Weinheim 2013, ISBN 978-3-527-33247-2, 21.1.2 Die Reaktionsgeschwindigkeit.
- ↑ Carl Heinz Hamann, Dirk Hoogestraat, Rainer Koch: Grundlagen der Kinetik: Von Transportprozessen zur Reaktionskinetik. Springer, Berlin,
Heidelberg 2017, ISBN 978-3-662-49392-2, 4.2.3.4 Enzymatische Katalyse, S. 167 ff.,
doi: 10.1007/978-3-662-49393-9.
- ↑ Hochspringen nach: a b M. Dieter Lechner: Einführung in die Kinetik: Chemische Reaktionskinetik
und Transporteigenschaften. Springer, Berlin / Heidelberg 2018, ISBN 978-3-662-57454-6, 3.1.5 Reaktionen mit einer Pseudoordnung, S. 13 f.,
doi: 10.1007/978-3-662-57455-3.
- ↑ Hochspringen nach: a b Marcus Elstner: Physikalische Chemie I: Thermodynamik und Kinetik. Springer,
Berlin / Heidelberg 2017, ISBN 978-3-662-55363-3, 17.3 Methoden zur Bestimmung der Reaktionsordnung, S. 298 f.,
doi: 10.1007/978-3-662-55364-0.
- ↑ M. Dieter Lechner: Einführung in die Kinetik: Chemische Reaktionskinetik und Transporteigenschaften. Springer Berlin Heidelberg,
Berlin, Heidelberg 2018, ISBN 978-3-662-57454-6, 3.1 Konzentration in Abhängigkeit von der Zeit, S. 9 ff.,
doi: 10.1007/978-3-662-57455-3.
- ↑ Hochspringen nach: a b Carl Heinz Hamann, Dirk Hoogestraat, Rainer Koch: Grundlagen der Kinetik: Von
Transportprozessen zur Reaktionskinetik. Springer, Berlin, Heidelberg 2017, ISBN 978-3-662-49392-2, 4.2.1.2.3 Die Bestimmung von Reaktionsordnungen, S. 117 ff.,
doi: 10.1007/978-3-662-49393-9.
- ↑ M. Dieter Lechner: Einführung in die Kinetik: Chemische Reaktionskinetik und Transporteigenschaften. Springer, Berlin, Heidelberg 2018,
ISBN 978-3-662-57454-6, 3.2 Methode der Anfangsgeschwindigkeit, S. 14 f.,
doi: 10.1007/978-3-662-57455-3.
- ↑ M. Dieter Lechner: Einführung in die Kinetik: Chemische Reaktionskinetik und Transporteigenschaften. Springer Berlin Heidelberg, Berlin,
Heidelberg 2018, ISBN 978-3-662-57454-6, 3.3 Halbwertszeit, S. 16 f.,
doi: 10.1007/978-3-662-57455-3.
- ↑ James H. Espenson: Chemical kinetics and reaction mechanisms (= MacGraw-Hill series in advanced chemistry). 2. Auflage. McGraw Hill, New York, NY 1987, ISBN 978-0-07-113949-6, S. 30–31.
- ↑ Carl Heinz Hamann, Dirk Hoogestraat, Rainer Koch: Grundlagen der Kinetik: Von Transportprozessen zur Reaktionskinetik. Springer, Berlin,
Heidelberg 2017, ISBN 978-3-662-49392-2, 4.2.1.2.2 Gebrochene und formale Reaktionsordnungen, S. 116 ff.,
doi: 10.1007/978-3-662-49393-9
( springer.com).
- ↑ John H. Sullivan: Mechanism of the Bimolecular Hydrogen—Iodine Reaction. In: The Journal of Chemical
Physics. Band 46, Nr. 1, 1. Januar 1967,
ISSN 0021-9606, S. 73–78,
doi: 10.1063/1.1840433
( aip.org [abgerufen am 17. Dezember 2025]).
- ↑ Keith J. Laidler: Chemical Kinetics. 3. Auflage. HarperCollins, New York 1998, ISBN 978-0-06-043862-3, 8.4.3 Hydrogen—Iodine Reaction, S. 298 f.
- ↑ M. Dieter Lechner: Einführung in die Kinetik: Chemische Reaktionskinetik und Transporteigenschaften. Springer, Berlin / Heidelberg 2018,
ISBN 978-3-662-57454-6, 5.7.3.1 Radikalpolymerisation, S. 67 ff.,
doi: 10.1007/978-3-662-57455-3
( springer.com).
- ↑ James H. Espenson: Chemical kinetics and reaction mechanisms (= MacGraw-Hill series in advanced chemistry). 2. Auflage. McGraw Hill, New York, NY 1987, ISBN 978-0-07-113949-6, S. 34, 60.
- ↑ Antonio E. Mucientes, María A. de la Peña: Ruthenium(VI)-Catalyzed Oxidation of Alcohols by Hexacyanoferrate(III): An Example of Mixed Order. In:
Journal of Chemical Education. Band 83, Nr. 11,
November 2006, S. 1643,
doi: 10.1021/ed083p1643.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 22.01. 2026