Glycoside
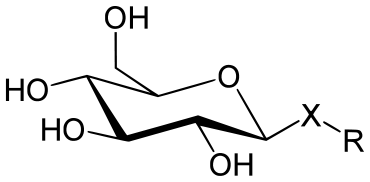
Glycoside, auch Glykoside, sind organische chemische Verbindungen der allgemeinen Struktur R–O–Z. Dabei ist ein Alkohol (R–OH) über eine glycosidische Bindung mit einem Zucker (Z) verbunden. Glycoside sind somit Vollacetale von Zuckern. Der Alkohol OR kann sowohl ein anderer Zucker als auch eine beliebige andere Hydroxyverbindung sein, aber kein Acylrest.
Liegt statt eines Acetals ein Thioacetal R–S–Z oder Selenoacetal R–Se–Z vor, spricht man von einem Thioglycosid bzw. Selenoglycosid.
N-Glycosyl-Verbindungen R–NR’–Z heißen dagegen Glycosylamine beziehungsweise Aminozucker. C-Glycosylverbindungen R–CR’R’’–Z sind Glycosylderivate. Im Laborjargon wird stattdessen oft nur „C-Glycosid“ bzw. „N-Glycosid“ gesagt, das ist jedoch irreführend, da sich Glycosylamine und Glycosyle chemisch unterschiedlich verhalten.
Der Zuckerteil Z wird allgemein als Glycon (Glykon) bezeichnet. Wenn es sich bei R–OH nach IUPAC-Nomenklatur um einen Nichtzucker handelt, wird es Aglycon (Aglykon) genannt.
Die glycosidische Bindung
Die chemische Bindung zwischen dem anomeren Kohlenstoffatom eines Zuckers und dem Heteroatom des Aglycons oder mit einem zweiten Zucker wird als glycosidische Bindung bezeichnet (siehe Abbildung). Glycoside sind auch Stoffe mit Bindungen zu anderen Heteroatomen wie Schwefel, Selen, Stickstoff und Phosphor. Die glycosidische Bindung ist hydrolytisch spaltbar, wobei das Reaktionsgleichgewicht auf Seiten der Spaltungsprodukte liegt. Die Bindung ist kinetisch aber recht stabil. Sie wird mit geringem Energieaufwand unter Wasserabspaltung durch eine Kondensationsreaktion gebildet. In der Natur erfolgt die als Glycosylierung bezeichnete Bildung enzymatisch über ein aktiviertes Saccharid, im Labor durch spezielle Aktivierungsmethoden oder durch Reaktion eines Zuckers mit einem großen Überschuss des Alkohols unter Säurekatalyse.
Bei einem Glycosid liegt die Aldehydfunktion der Aldosen (z.B. Glucose) oder die Ketofunktion der Ketosen (z.B. Fructose) als cyclisches Vollacetal vor. Ein Acetal ist das Kondensationsprodukt aus einem Aldehyd oder Keton und einem oder zwei Alkoholen (Halbacetal bzw. Vollacetal). Vollacetale sind stabil gegen basische und neutrale bis schwach saure wässrige Lösungen, hydrolysieren jedoch in Gegenwart starker Säuren.
Stereochemie und Nomenklatur
Durch die Bildung des glycosidischen Vollacetals wird die prochirale Carbonylfunktion chiral, d.h., es bildet sich ein neues Stereozentrum, das sogenannte anomere Kohlenstoffatom oder anomere Zentrum. Die beiden resultierenden Diastereomere (Anomere) werden als α- bzw. β-Glycosid bezeichnet. Die Glycoside werden durch Anhängen der Endung -id an den Wortstamm des Glycons benannt, z.B. Fructosid für ein Glycosid der Fructose oder Glucosid für ein Glycosid der Glucose.
Bildet der Zucker einen Fünfring, so handelt es sich um ein Glycofuranosid (abgeleitet von Furan). Bildet er einen Sechsring, so spricht man von einem Glycopyranosid (abgeleitet von Pyran).
- Siehe auch Glycosidische Bindung#Nomenklatur und Beispiele
- Definition α und β: Glycosidische Bindung
Glycosylierung und Hydrolyse
Was die Peptidbindung bei den Aminosäuren ist, ist die glycosidische Bindung bei den Kohlenhydraten, da sie durch eine leicht reversible Kondensationsreaktion eine stabile Verknüpfung zu anderen Zuckern oder den verschiedensten Alkoholen ermöglicht. Dadurch ergibt sich für die Kohlenhydrate, die mit Abstand den größten Teil der Biomasse ausmachen, eine gewaltige Strukturvielfalt. Sowohl die enzymatische als auch die nicht-enzymatische Synthese eines Glycosids wird als Glycosylierung bezeichnet.
Enzyme
In biologischen Systemen werden Glycoside durch Glycosidasen zum freien Zucker und dem aglyconischen Alkohol hydrolysiert. Diese Glycosidasen sind mehr oder weniger spezifisch für bestimmte Zucker und eine der anomeren Formen und Ringgrößen. So kann eine α-Mannopyranose-spezifische Glycosidase, eine α-Mannosidase, kein Galactosid hydrolysieren. Eine α-Galactosidase kann aber nicht nur α-Galactopyranoside spalten, sondern manchmal auch ähnliche Glycoside, wie zum Beispiel β-Arabinopyranoside. Glycosyltransferasen hingegen sind hochspezifische Enzyme, die die Übertragung aktivierter Kohlenhydrate (UDP-Zucker) auf einen Alkohol unter Ausbildung einer glycosidischen Bindung katalysieren.
Chemische Synthesen
Die Ausbildung glycosidischer Bindungen ist eine der wichtigsten Bestandteile der Kohlenhydratchemie. Es gibt mittlerweile eine Vielzahl verschiedener enzymatischer und nicht-enzymatischer Glycosylierungsmethoden, die im Labor bis hin zur industriellen Produktion eingesetzt werden.
Fischer-Glycosylierung: Einfache Alkylglycoside und Alkylpolyglycoside
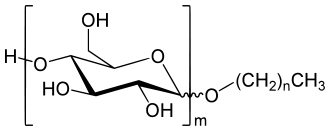
Die glycosidische Bindung ist stabil gegenüber einem basischen bis schwach sauren Milieu, aber empfindlich gegenüber stark sauren Bedingungen, ggf. unter Energiezufuhr oder erhöhtem Druck. Die Nachbarschaft des Ringsauerstoffs begünstigt die Solvolyse durch Stabilisierung des gebildeten cyclischen Carboxoniumions. In wässriger Lösung führt dies zur Hydrolyse eines Glycosids zum Aglycon und dem freien Zucker. In einer alkoholischen Lösung wird stattdessen das zum Alkohol korrespondierende Alkylglycosid gebildet, z.B.:

| Monosaccharid | Me-α-pyr | Me-β-pyr | Me-α-fur | Me-β-fur |
|---|---|---|---|---|
| Glucose | 66 | 32,5 | 0,6 | 0,9 |
| Mannose | 94 | 5,3 | 0,7 | - |
| Galactose | 58 | 20 | 6 | 16 |
| Arabinose | 24 | 47 | 22 | 7 |
Es ist zu beachten, dass es sich bei dieser nach Emil Fischer benannten
Fischer-Glycosylierung um eine komplexe Gleichgewichtsreaktion handelt, mit dem
freien Zucker als thermodynamischen Produkt. Um eine quantitative Ausbeute an
Alkylglycosid zu erhalten, wird wasserfreier Alkohol verwendet, meist mit
HClg oder stark saurem Ionentauscher als
Katalysator. Das bei der Reaktion entstehende Wasser kann z.B. in einer Soxhlet-Apparatur durch
Molekularsieb dynamisch
entzogen werden.
Auf der Glycosid-Seite entstehen sowohl die α- als auch
β-Glycopyrano- und furanoside. Die Gleichgewichtsmischung der Glycoside bei
Wasserausschluss ist für jedes Saccharid individuell verschieden, meist sind
jedoch die α-Glycopyranoside das Hauptprodukt (siehe Tabelle). Durch
Kristallisation können diese dann oftmals sehr rein erhalten werden. Dies
funktioniert für einfache Alkohole bis zum Butanol sehr gut. Wenn das gewünschte
Aglycon jedoch unpolarer ist, bekommt man ein Phasenproblem. Der Zucker ist im
Alkohol nicht mehr löslich und die Reaktion kann nicht stattfinden, bzw. es sind
sehr hohe Temperaturen oder Druck erforderlich. Seit einigen Jahren haben die Alkylpolyglycoside
(APG) im Sinne der nachwachsenden
Rohstoffe eine industrielle Bedeutung als Tenside,
insbesondere in hochwertigen Kosmetika erlangt. Da hierfür die Glycoside
langkettiger und unpolarer Fettalkohole interessant sind, hat man das
beschriebene Phasenproblem. Daher wird ausgehend von Stärke zunächst eine Butanolyse durchgeführt und die
so entstandenen Butylpolyglycoside mit den Fettalkoholen umglycosyliert:
![{\mathrm {(Glucose)_{{n}}+Butanol\,{\xrightarrow {[H^{+}]}}\,Butylpolyglucoside\,{\xrightarrow {[H^{+}],\,Fettalkohole}}\,Alkylpolyglucoside}}](/svg/f96fc02028f543bf01b73f665817aa006ac720b6.svg)
Anomerer Effekt
Die erhöhte thermodynamische Stabilität der axialen anomeren Stellung (meistens α) wird als anomerer Effekt bezeichnet und ist Gegenstand kontroverser Diskussionen. Wie stark der anomere Effekt ausgeprägt ist, hängt von der Konfiguration des Zuckers und u. a. von der Polarität des Lösungsmittels ab. Er spielt in der Kohlenhydratchemie eine bedeutende Rolle und kann in der Synthese ausgenutzt werden.
Aktivierung geschützter Glycosyldonoren
In einer Glycosylierungsreaktion wird der Zucker als Glycosyldonor und das Aglycon als Glycosylakzeptor bezeichnet. Ziel einer solchen Synthese ist in der Regel die Einführung eines empfindlichen Aglycons oder die Synthese von Oligosacchariden. Die einfachen Alkylglycoside werden meist durch Fischer-Glycosylierung (nach Kristallisation hauptsächlich oder rein α) bzw. aus peracetylierten Zuckern (β, siehe unten) erhalten.
- Schutzgruppen: Um eine selektive Glycosylierung zu erhalten, muss wegen der hohen Funktionalisierung der Saccharide ein erheblicher Mehraufwand betrieben werden. Es wird eine Vielzahl von Schutzgruppen zur Maskierung der Hydroxygruppen verwendet. Die wichtigsten Schutzgruppen sind Acetate, Benzoate und Benzylether.
- Nachbargruppeneffekt: 2-O-Acylierte Glycosyldonoren ergeben
selektiv β-Glycoside (bzw. α bei u.a. Mannose), da der
Ester-Carbonylsauerstoff die α-Position abschirmt. Dagegen ergeben
2-O-alkylierte Donoren überwiegend bzw. unter sehr speziellen
Bedingungen ausschließlich die thermodynamisch günstigeren α-Glycoside (gilt
ebenso für Mannose).
Als störend kann sich beim Nachbargruppeneffekt die Ausbildung eines Orthoesters als Konkurrenzprodukt zum Glycosid erweisen.
Um einen vollwertigen Glycosyldonor zu erhalten, muss eine geeignete Abgangsgruppe in der anomeren Position eingeführt werden. An dieser Stelle sollen nur einige wichtige Donoren und Ihre Aktivierung in Gegenwart des Alkohols kurz beschrieben werden:
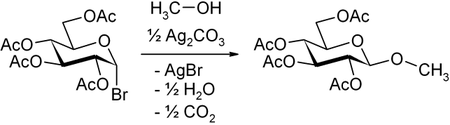
- Zuckerhalogenide: Eine der bekanntesten Glycosylierungsmethoden ist die Aktivierung von Zuckerhalogeniden durch Silbersalze, bekannt als Koenigs-Knorr-Methode, zum Beispiel Acetobromglucose durch Silbercarbonat (man beachte, dass hier durch die benachbarte 2-OAc-Gruppe selektiv β entsteht):
- Trichloracetimidate: Zucker–O–(C=NH)–CCl3 werden durch katalytische Mengen Lewis-Säure aktiviert.
- Thioglycoside: Zucker-S-R werden u. a. durch elektrophile Reagenzien wie Br+ aktiviert.
- 1-O-Acetate: Zucker-OAc werden durch einen Überschuss Lewis-Säure aktiviert. Sie werden als peracetylierte Verbindungen zur Synthese von Alkyl-β-glycosiden und -thioglycosiden, sowie geschützten 1-OH-freien Zuckern und Zuckerhalogeniden verwendet.
Biologie und Pharmazie
Glycoside sind in der Natur weit verbreitet. Sie haben eine große Bandbreite an biologischen Funktionen. Bei der Unterscheidung nach der Zugehörigkeit des Aglycons zu einer bestimmten chemischen Stoffgruppe entstehen umfangreiche Unterklassen, die sich oft in der Toxizität, der Eignung als Arzneistoffe oder durch sonstige Eigenschaften ähneln. In der Biochemie und der Pharmakologie ist dies die gängigste Klassifizierung.
Im menschlichen Körper werden polare und unpolare alkoholische Giftstoffe oft durch Bindung an Glucuronsäure als Glucuronide wasserlöslich gemacht und ausgeschieden.
Einige spezielle Glycoside sind sekundäre Pflanzenstoffe. Die Synthese von Glycosiden ermöglicht es der Pflanze u.a., toxische Stoffe in nicht-toxischer Form zu speichern. Dabei wird das Glycosid z.B. in einer Vakuole gespeichert, was der Kompartimentierung von der jeweiligen Glycosidase dient. Kommen das Glycosid und die zugehörige Glycosidase, z.B. durch Zerstörung der Pflanzenzelle zusammen, wird das Glycosid hydrolytisch gespalten und der Giftstoff wird freigesetzt und kann seine Wirkung entfalten.
In ihrer Wirkung als Arzneistoffe bzw. ihrer Toxikologie sind Glycoside sehr unterschiedlich. Sie werden in Biochemie und Pharmazie je nach Aglycon in folgende Untergruppen aufgeteilt:
Anthocyanglycoside
Die Anthocyanglycoside bilden eine Gruppe von in vielen Pflanzen als Farbstoffe vorkommender Verbindungen.
Cumaringlycoside
Die Cumaringlycoside (zum Beispiel Rutarin) leiten sich von dem als Duftstoff eingesetzten Cumarin ab. Viele dieser Glycoside besitzen eine pharmakologische Wirkung.
Cyanogene Glycoside
Cyanogene Glycoside spalten – wie ihr Name ausdrückt – bei ihrer Zersetzung die stark toxische Blausäure HCN ab.
Flavonoide
Bei der Gruppe der Flavonoide ist das Aglycon ein Flavon. Diese große Gruppe beinhaltet ähnlich wie die Anthocyanglycoside viele pflanzliche Farbstoffe, die als natürliche Polyphenole ebenfalls pharmakologisch wirksam sind. Beispiele sind das Hesperidin, Naringin und Rutin.
Herzglycoside
Herzglycoside sind etwa 300 Stoffe, die pharmakologisch inotrop auf die Herzmuskulatur wirken. Sie kommen in einigen Pflanzen wie den Fingerhüten Digitalis oder dem Maiglöckchen Convallaria majalis vor und bestehen aus drei relativ seltenen Desoxyzuckern und einem Steroidalkohol als Aglycon. Heute besitzen nur noch Digoxin und Digitoxin klinische Bedeutung. Wegen ihrer speziellen Eigenschaften werden sie von den verwandten Saponinen unterschieden. Wie man inzwischen weiß, kann unser Körper selbst herzwirksame Glycoside aus Cholesterin herstellen, die Bedeutung endogen synthetisierter Herzglycoside ist jedoch noch unbekannt.
Iridoidglycoside
Iridoidglycoside wie Aucubin und Catalpol dienen der Abwehr von Fressfeinden der Pflanzen.
Phenolglycoside oder Phenylglycoside
Phenylglycoside (auch Phenolglycoside) sind eine Gruppe von Glycosiden, bei denen ein Phenol über eine glycosidische Bindung mit einem Kohlenhydrat verknüpft ist. Da viele Anthrachinone und Flavonoide ebenfalls Phenole sind, überlappen diese Gruppen teilweise. Phenylglycoside sind in der Natur weit verbreitet; hierzu gehören z.B.:
- Aesculin
- bestimmte Anthraglycoside
- Anthocyane
- Arbutin
- Coniferin
- Hesperidin
- Naringin
- Phlorizin
- Salicin: Ein Beispiel ist das in der Weide Salix vorkommende Salicin. Es entsteht aus dem Glycon D-Glucose und Salicylalkohol (auch Saligenin). Im menschlichen Organismus wird der Stoff hydrolysiert und es entstehen Glucose und Salicylalkohol, der in der Leber zu Salicylsäure metabolisiert wird.
- Scutellarin
- Vanillinglucosid
Saponine
Saponine (lat. „Sapo“ = „Seife“) bilden beim Mischen und Schütteln mit Wasser seifenartige Mischungen. Das jeweilige Aglycon zählt zur Klasse der Sapogenine, das meist mit D-Glucose oder D-Galactose verbunden ist. Sapogenine sind entweder Steroide, Steroidalkaloide (stickstoffhaltige Steroide) oder Triterpene. Vielen Pflanzen dienen Saponine wie Digitonin oder Solanin als Defensiv- bzw. Abwehrstoffe.
Senfölglycoside
Senfölglycoside oder Glucosinolate sind Glycoside, die über ein Schwefelatom verbrückt sind, also S-Glycoside oder Thioglycoside. Zusätzlich enthalten diese auch Stickstoff und verleihen vielen Kreuzblütlern und Kaperngewächsen wie Rettich, Senf, Kresse und Kohl einen bitteren und scharfen Geschmack. Bei der Verletzung des Pflanzengewebes werden die Senfölglycoside in teils toxische Produkte abgebaut.
Klassischer Nachweis
Kohlenhydrate (Zucker) sind Polyhydroxycarbonylverbindungen, d.h., sie haben mehrere funktionelle Alkoholgruppen und liegen daher meist als energetisch sehr günstiges cyclisches Halbacetal vor, reagieren also mit sich selbst unter Ringbildung. Das ehemalige Carbonylsauerstoffatom bildet eine exocyclische OH-Gruppe, der Sauerstoff der Hydroxygruppe bildet den endocyclischen Ringsauerstoff. Da die Halbacetalform der Aldosen in wässriger Lösung mit der offenkettigen aldehydischen Form im Gleichgewicht liegt, reduziert eine Glucose-Lösung Fehlingsche Lösung. Zucker bilden mit den verschiedensten Alkoholen R-OH ein cyclisches Vollacetal, bei dem statt einer exocyclischen Hydroxygruppe ein exocyclischer Substituent OR vorliegt. Ein solches Vollacetal ist in wässriger Lösung stabil und reduziert Fehlingsche Lösung daher nicht, es sei denn das Aglycon selbst wirkt reduzierend, z.B. wenn es sich wiederum um einen Zucker handelt. In stark saurer wässriger Lösung werden Glycoside zu einem oder mehreren Monosaccharid(en) und dem Alkohol gespalten. Das klassische Nachweis-Kriterium für ein Glycosid ist daher die Beständigkeit gegen Fehlingsche Lösung ohne vorherige Hydrolyse und Reduktion von Fehlingscher Lösung nach saurer Hydrolyse. Bei Vorliegen eines reduzierenden Aglycons, z.B. einem weiteren Zucker, greift dieses Kriterium allerdings nicht. Da erste Untersuchungen pharmazeutischer pflanzlicher Formulierungen (Formulierung = Darreichungsform) mit diesen klassischen Untersuchungsmethoden erfolgten, werden in der Pharmazie die unten angegebenen pflanzlichen Wirkstoffe als Glycoside bezeichnet, obwohl die Glycoside eine weit über diese kleine Gruppe hinausgehende Bedeutung haben.
Literatur
- Jochen Lehmann, Kohlenhydrate: Chemie und Biologie, 2. neu bearb. und erw. Aufl., Stuttgart · New York, Thieme 1996.


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 17.04. 2026