Van-der-Waals-Gleichung
Die Van-der-Waals-Gleichung ist eine Zustandsgleichung für Gase, mit der das Verhalten realer Gase in besserer Annäherung beschrieben werden kann als mit der Allgemeinen Gasgleichung für das Ideale Gas. Die Van-der-Waals-Gleichung enthält, über die allgemeine Gasgleichung hinausgehend, zwei Parameter für die abstoßenden und die anziehenden Kräfte zwischen den Gasteilchen. Diese sind charakteristisch für das jeweilige Gas. Damit führt sie zu einem einfachen und näherungsweise quantitativen Verständnis der Verflüssigung und vieler weiterer Eigenschaften, in denen die realen Gase vom idealen Gas abweichen. Die Gleichung wurde 1873 durch Johannes Diderik van der Waals aufgestellt, wofür er 1910 den Nobelpreis für Physik erhielt.
Die Gleichung und ihre Interpretation
Gleichung
| Gas | Kohäsionsdruck a in (kPa·dm6)/mol2 = 10−3·(Pa·m6)/mol2 = 10−3·(J·m3)/mol2 |
Kovolumen b in cm3/mol = 10−6·m3/mol | |
|---|---|---|---|
| Helium (He) | 3,45 | 23,7 | |
| Neon (Ne) | 21,3 | 17,1 | |
| Argon (Ar) | 136,3 | 32,2 | |
| Wasserstoff (H2) | 24,7 | 26,6 | |
| Stickstoff (N2) | 140,8 | 39,1 | |
| Sauerstoff (O2) | 137,8 | 31,8 | |
| Luft (80 % N2, 20 % O2) | 135,8 | 36,4 | |
| Kohlendioxid (CO2) | 363,7 | 42,7 | |
| Wasser (H2O) | 557,29 | 31 | |
| Chlor (Cl2) | 657,4 | 56,2 | |
| Ammoniak (NH3) | 422,4 | 37,1 | |
| Methan (CH4) | 225 | 42,8 | |
| Benzol (C6H6) | 52,74 | 304,3 | |
| Decan (C10H22) | 37,88 | 237,4 | |
| Octan (C8H18) | 18,82 | 119,3 | |
| Die hier integrierten Literaturwerte
basieren auf „technisch reinen“ Stoffen. Sie gelten nicht für Flüssigkeiten, sondern in der Gasphase. | |||
Die Van-der-Waals-Gleichung lautet
 bzw.
bzw.

oder gleichbedeutend

Die Formelzeichen stehen für folgende Größen:
 – Druck
des Gases
– Druck
des Gases – Temperatur
– Temperatur – molares
Volumen (
– molares
Volumen ( mit Volumen
mit Volumen  und Stoffmenge
und Stoffmenge  des Gases)
des Gases) – Universelle
Gaskonstante (wie beim idealen Gas)
– Universelle
Gaskonstante (wie beim idealen Gas)
 – Kohäsionsdruck(-parameter)
– Kohäsionsdruck(-parameter) – Kovolumen
– Kovolumen
Kohäsionsdruck(-parameter)
und Kovolumen
werden auch als Van-der-Waals-Konstanten des betreffenden Gases
bezeichnet (siehe Beispiele in der Tabelle). Für das ideale Gas gilt  und
und  .
.
Durch Einführung der Stoffmenge
kann die Van-der-Waals-Gleichung auch dargestellt werden als

Die Van-der-Waals-Gleichung beschreibt sowohl die Gas- als auch die Flüssigphase qualitativ richtig, ist jedoch für quantitative Anwendungen oft zu ungenau. Infolgedessen können die van der Waals-Parameter eines Gases davon abhängen, in welchem Bereich von Zuständen sie gewonnen wurden. Genauer sind z.B. die Redlich-Kwong-Gleichung oder die Zustandsgleichung von Soave-Redlich-Kwong, beides halbempirische modifizierte Van-der-Waals-Gleichungen.
Es gibt auch empirische Zustandsgleichungen, wie z.B. die Benedict-Webb-Rubin-Gleichung.
Interpretation im Rahmen der kinetischen Gastheorie
Die Zustandsgleichung des idealen Gases

lässt sich in der kinetischen Gastheorie unter der Annahme punktförmiger Teilchen ohne gegenseitige Kräfte begründen (Herleitung). Im Vergleich dazu ist beim Van-der-Waals-Gas der Druck
- verringert durch Abzug des Kohäsionsdrucks
(oder Binnendrucks)

- erhöht um einen Faktor
 durch die effektive Verringerung des molaren Volumens um das Kovolumen
.
durch die effektive Verringerung des molaren Volumens um das Kovolumen
.
Auch diese Modifikationen lassen sich mit der kinetischen Gastheorie anschaulich interpretieren.
Kohäsionsdruck
Der Van-der-Waals-Parameter
trägt der Tatsache Rechnung, dass Gasmoleküle einander anziehen. Das wird im
Modell des idealen Gases ignoriert, ist aber unter anderem für die Möglichkeit
der Verflüssigung ausschlaggebend (daher die Bezeichnung Kohäsionsdruck). Diese
unter dem Namen Van-der-Waals-Kräfte
zusammengefassten anziehenden Kräfte sind letztlich elektromagnetischer Natur,
haben aber sehr viel kleinere Reichweite. Sie beruhen darauf, dass Moleküle,
sogar solche ohne permanentes Dipolmoment, einander elektrisch polarisieren
können, besonders markant bei zwei gleichen Molekülen. Diese Kräfte verringern
den Druck auf die Behälterwand, weil sie für die Moleküle nahe der Wand
vorwiegend nach innen gerichtet sind (daher die Bezeichnung Binnendruck). Pro
Teilchen ist ihre Stärke proportional zur Dichte der übrigen Teilchen, für alle
Teilchen zusammen also proportional zum Quadrat der Teilchendichte. Daher
ist der negative Gesamtbeitrag zum Druck umgekehrt proportional zu  .
.
Kovolumen
Durch den Van-der-Waals-Parameter
wird berücksichtigt, dass die Gasmoleküle keine Massenpunkte sind, wie im Modell
des idealen Gases angenommen. Aufgrund ihrer endlichen Größe ist die mittlere freie
Weglänge verkürzt und die Zahl der Stöße gegen die Wand, die den Druck
erzeugen, wie auch die der Stöße untereinander, größer als für punktförmige
Teilchen. Die Erhöhung der Stoßzahl kann durch eine scheinbare Verringerung des
Volumens parametrisiert werden. Anschaulich gesagt können die
Teilchenmittelpunkte sich nur bis zum doppelten Radius annähern, wodurch
effektiv aber nicht das Achtfache, sondern nur das Vierfache ihres Eigenvolumens
gesperrt wird (denn hier sind nur die möglichen Teilchenpaare zu zählen). Daraus
ergibt sich, dass das Kovolumen
etwa die Größe des 4fachen Eigenvolumens der Moleküle hat. Das Kovolumen
entspricht auch etwa dem Volumen des verflüssigten Gases.
Begründung in der Statistischen Mechanik
Eine systematische Begründung der van der Waals-Gleichung und ihrer Parameter
und
wird in der statistischen
Mechanik gegeben. Dort wird die Zustandsgleichung im Rahmen einer Reihenentwicklung
nach der Teilchendichte berechnet (siehe Virialentwicklung).
Dabei ergibt sich für ein klassisches Gas die Zustandsgleichung des idealen
Gases, wenn man diese Reihenentwicklung nach dem ersten Glied abbricht, und die
van der Waals-Gleichung (in entsprechender Näherung), wenn man das folgende zweite
Glied mit berücksichtigt.
Isothermen im p-V-Diagramm
Die Isotherme nach der nach
aufgelösten Van-der-Waals-Gleichung

wird im p-V-Diagramm
durch eine Kurve dargestellt, die die Differenz einer einfachen Hyperbel und
einer quadrierten Hyperbel ist, wobei die einfache Hyperbel um
verschoben ist.
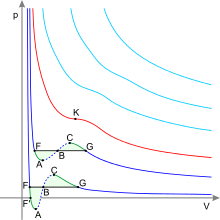
und Maxwell-Konstruktion
Für hohe Temperaturen
und geringe Dichte (d.h. große )
nähert sie sich der einfachen Hyperbel entsprechend der Gleichung für ideale
Gase.
Unterhalb einer kritischen
Temperatur  (rot markierte Isotherme im Bild) treten jedoch ein Maximum und ein Minimum auf
(Punkte C und A), so dass bei einer isothermen Kompression von C nach A sowohl
das Volumen als auch der Druck abnehmen würden. Dieser Bereich ist daher
instabil und kommt in der Natur auch nicht in metastabiler Form vor. Stattdessen
verläuft die Isotherme in diesem Volumenbereich isobar, d.h. im --Diagramm
als horizontale Linie (schwarz). Geht man von einem großen Anfangsvolumen aus
durch eine isotherme Kompression zu kleineren Volumina, dann erhöht sich
zunächst der Druck gemäß der van der Waals-Isotherme, bleibt aber ab einem
bestimmten Wert (Punkt G in der Abbildung) konstant, auch wenn das Volumen
weiter verkleinert wird. Ab diesem Punkt findet – wenn die Temperatur weiterhin
durch Wärmeabfuhr konstant gehalten wird – bei konstantem Druck die Kondensation des Gases zur
Flüssigkeit statt. Längs der schwarzen Linie befinden sich beide
Aggregatzustände in Koexistenz, wobei sich nur ihr Mengenverhältnis ändert.
Dabei behält der gasförmige Anteil die Dichte, die er am Punkt G hatte, und der
flüssige die Dichte des Punktes F. Der Bereich der Koexistenz auf dieser
Isotherme endet, wenn das isobare Stück wieder auf die van der Waals-Kurve
trifft (Punkt F in der Abbildung). Ab dieser Stelle steigt der Druck bei
weiterer Verringerung des Volumens steil an, was für die geringe
Kompressibilität einer reinen Flüssigkeit charakteristisch ist. Zusammen bilden
diese isobaren Bereiche der Isothermen aller Temperaturen bis zur kritischen
Temperatur das Koexistenzgebiet Gas-Flüssigkeit des betrachteten Gases.
(rot markierte Isotherme im Bild) treten jedoch ein Maximum und ein Minimum auf
(Punkte C und A), so dass bei einer isothermen Kompression von C nach A sowohl
das Volumen als auch der Druck abnehmen würden. Dieser Bereich ist daher
instabil und kommt in der Natur auch nicht in metastabiler Form vor. Stattdessen
verläuft die Isotherme in diesem Volumenbereich isobar, d.h. im --Diagramm
als horizontale Linie (schwarz). Geht man von einem großen Anfangsvolumen aus
durch eine isotherme Kompression zu kleineren Volumina, dann erhöht sich
zunächst der Druck gemäß der van der Waals-Isotherme, bleibt aber ab einem
bestimmten Wert (Punkt G in der Abbildung) konstant, auch wenn das Volumen
weiter verkleinert wird. Ab diesem Punkt findet – wenn die Temperatur weiterhin
durch Wärmeabfuhr konstant gehalten wird – bei konstantem Druck die Kondensation des Gases zur
Flüssigkeit statt. Längs der schwarzen Linie befinden sich beide
Aggregatzustände in Koexistenz, wobei sich nur ihr Mengenverhältnis ändert.
Dabei behält der gasförmige Anteil die Dichte, die er am Punkt G hatte, und der
flüssige die Dichte des Punktes F. Der Bereich der Koexistenz auf dieser
Isotherme endet, wenn das isobare Stück wieder auf die van der Waals-Kurve
trifft (Punkt F in der Abbildung). Ab dieser Stelle steigt der Druck bei
weiterer Verringerung des Volumens steil an, was für die geringe
Kompressibilität einer reinen Flüssigkeit charakteristisch ist. Zusammen bilden
diese isobaren Bereiche der Isothermen aller Temperaturen bis zur kritischen
Temperatur das Koexistenzgebiet Gas-Flüssigkeit des betrachteten Gases.
Die Lage des horizontalen Stücks, also der Sättigungsdampfdruck des verflüssigten Gases bei der Temperatur der betrachteten Isotherme, kann daraus bestimmt werden, dass die beiden Flächenstücke zur van der Waals-Kurve hin (lindgrün in der Abbildung) gleich groß sein müssen. James Clerk Maxwell, der diese Maxwell-Konstruktion einführte, begründete sie mit der Energieerhaltung bei einem isothermen reversiblen Kreisprozess längs der Van-der-Waals-Schleife, die die beiden Flächen berandet. Jedoch krankt diese Begründung daran, dass der Kreisprozess wegen der Instabilität zwischen A und C gar nicht wirklich stattfinden kann. Eine bessere Begründung beruht darauf, dass die Zustände auf dem betreffenden Teil der Van-der-Waals-Isotherme instabil sind und von alleine in Zustände auf der Maxwell-Geraden übergehen, weil diese die geringste bei gleicher Temperatur und gleichem Druck mögliche Freie Enthalpie haben. Die von der Van-der-Waals-Gleichung beschriebene, in sich homogene Materie zerfällt dabei spontan in zwei Teile verschiedener Dichte. Der so bestimmte Dampfdruck hängt von der Temperatur ab, wie nach der Clausius-Clapeyron-Gleichung gefordert.
Kondensation und Verdampfung sind Phasenübergänge, die bei genügend schnellen Zustandsänderungen unter Umständen erst verzögert einsetzen. Daher können die Zustände auf den Kurvenstücken der van der Waals-Isotherme links vom Minimum (Punkte F-A) und rechts vom Maximum (Punkte C-G) zum Teil kurzzeitig erreicht werden, wenn das Volumen des reinen Gases schnell verringert oder das der reinen Flüssigkeit schnell vergrößert wird, oder durch andere geeignete Prozesse, die nicht-isotherm sind wie z.B. Siedeverzug. Diese Zustände stellen metastabile Zustände mit homogener Dichte dar, die bei Eintreten der Dampfbildung bzw. der Kondensation in einen passenden Mischzustand zerfallen. Z.B. konnten Wassertropfen unter Atmosphärendruck kurzzeitig bis 279 °C erhitzt werden, bis sie explosionsartig verdampften. Dabei bestätigte sich näherungsweise der Verlauf nach der van-Waals-Gleichung.
Kritischer Punkt
Bei einer Temperatur oberhalb der kritischen Temperatur zeigt die van der
Waals-Isotherme im  -Diagramm
überall negative Steigung. Es kann daher kein Bereich existieren, wo Volumen
(bzw. Dichte) variieren, die Temperatur aber gleich bleibt. Eine Koexistenz von
zwei Phasen verschiedener Dichte ist damit ausgeschlossen. Das stimmt mit allen
Beobachtungen an realen Gasen überein. Die kritische Temperatur
-Diagramm
überall negative Steigung. Es kann daher kein Bereich existieren, wo Volumen
(bzw. Dichte) variieren, die Temperatur aber gleich bleibt. Eine Koexistenz von
zwei Phasen verschiedener Dichte ist damit ausgeschlossen. Das stimmt mit allen
Beobachtungen an realen Gasen überein. Die kritische Temperatur  ist dadurch gegeben, dass ihre Isotherme durch den kritischen Punkt geht, wo sie
im -Diagramm
einen Wendepunkt
ist dadurch gegeben, dass ihre Isotherme durch den kritischen Punkt geht, wo sie
im -Diagramm
einen Wendepunkt  mit horizontaler Tangente hat: Am kritischen Punkt verschwindet die erste und
die zweite Ableitung des Drucks nach dem Volumen (bei konstanter Temperatur):
mit horizontaler Tangente hat: Am kritischen Punkt verschwindet die erste und
die zweite Ableitung des Drucks nach dem Volumen (bei konstanter Temperatur):

Daraus folgen:



Einige Beispiele für kritische Temperaturen: Wasser =374,12 °C,
Stickstoff =-146 °C,
Kohlenstoffdioxid =31 °C,
Helium-4 =5,2 K.
Nur unterhalb dieser Temperaturen lässt sich das betreffende Gas verflüssigen.
Aus den experimentell gefundenen Daten des kritischen Punktes lassen sich
umgekehrt die van der Waals-Konstanten
und
berechnen. Da es drei kritische Daten, aber nur zwei Van-der-Waals-Konstanten
gibt, ist das Gleichungssystem überbestimmt und hätte nur dann eine exakte
Lösung, wenn das Gas sich genau nach der Van-der-Waals-Gleichung verhielte. In
Tabellenwerken werden für die Van-der-Waals-Parameter oft die aus  gewonnenen Werte angegeben:
gewonnenen Werte angegeben:


Der Wert für  ist meist nur weniger genau bekannt, würde aber mit
ist meist nur weniger genau bekannt, würde aber mit  zu einem deutlich anderen Wert führen. Das drückt sich auch dann aus, wenn man
den Kompressionsfaktor
zu einem deutlich anderen Wert führen. Das drückt sich auch dann aus, wenn man
den Kompressionsfaktor
 berechnet. Für jedes ideale Gas ist in allen Zuständen
berechnet. Für jedes ideale Gas ist in allen Zuständen  .
Nach Van-der-Waals-Gleichung müsste unabhängig von den Parametern
und ,
also für jedes reale Gas gelten:
.
Nach Van-der-Waals-Gleichung müsste unabhängig von den Parametern
und ,
also für jedes reale Gas gelten:

Tatsächlich liegen die Werte für reale Gase relativ eng beisammen, sind aber noch weiter vom Wert 1 des idealen Gases entfernt als beim van der Waals-Modell. Beispiele:

In der relativ guten Übereinstimmung untereinander zeigt sich die Leistungsfähigkeit des Modells von van der Waals, in der Abweichung vom Modellwert dessen Näherungscharakter.
Reduzierte Form der van der Waals-Gleichung
Drückt man Druck, Temperatur und Molvolumen gemäß
 ,
,  ,
, 
durch die dimensionslosen reduzierten Zustandsgrößen  aus, dann heißt die Van-der-Waals-Gleichung:
aus, dann heißt die Van-der-Waals-Gleichung:

Diese Gleichung enthält keinen materialspezifischen Parameter und gilt daher
in identischer Form für alle Stoffe, die eine Van-der-Waals-Gleichung befolgen.
Dies ist ein Beispiel für das Theorem
der übereinstimmenden Zustände. Wenn zwei Substanzen dieselben Werte  annehmen, spricht man von übereinstimmenden/korrespondierenden Zuständen.
annehmen, spricht man von übereinstimmenden/korrespondierenden Zuständen.
Nach Potenzen des Volumens geordnet ergibt sich ein Polynom 3. Grades:

Der dimensionslose Kompressionsfaktor
 stellt einen Bezug zum Molvolumen eines idealen Gases her:
stellt einen Bezug zum Molvolumen eines idealen Gases her:

Damit lautet die Van-der-Waals-Gleichung:

Das stellt eine Gleichung 3. Grades für den Kompressionsfaktor dar:

Eine Näherung für kleine Drücke und/oder hohe Temperaturen ist:

Wärmeausdehnungskoeffizient
Der isobare Ausdehnungskoeffizient ergibt sich aus der Zustandsgleichung, wenn man sie in der oben zuerst angegebenen Form differenziert:

und dann  setzt. Es folgt
setzt. Es folgt

Für den Unterschied zu dem Ausdehnungskoeffizienten des idealen Gases  ,
der sich für
,
der sich für  ergibt, berechnet man
ergibt, berechnet man

Im Bereich normaler Temperaturen ist die Differenz für Gase wie Sauerstoff, Stickstoff, Luft positiv; diese Gase dehnen sich also etwas stärker aus als ein ideales Gas. Bei Wasserstoff und den Edelgasen ist es umgekehrt. Bei ihnen sind die Anziehungskräfte zwischen den Molekülen bzw. Atomen, und damit der Van-der-Waals-Parameter a, so klein, dass (bei nicht zu tiefer Temperatur) der Zähler negativ wird. Die Nullstelle des Zählers markiert auch die Zustände, bei denen der Joule-Thomson-Effekt zwischen Abkühlung und Erwärmung umschlägt (Inversionspunkt).
Innere Energie
Für ein Van-der-Waals-Gas ohne innere Freiheitsgrade gilt die kalorische Zustandsgleichung:

bzw. allgemeiner:  , wobei
, wobei  der volumenunabhängige Anteil der inneren Energie pro Teilchen ist.
der volumenunabhängige Anteil der inneren Energie pro Teilchen ist.
Sie hängt nicht nur von der kinetischen
Energie der Moleküle ab, sondern auch von der potentiellen
Energie der Kohäsionskräfte, gegeben durch den Parameter .


© biancahoegel.de
Datum der letzten Änderung: Jena, den: 28.05. 2024